„Nienaturalna ewolucja”: niepodważalne dowody na celowe i systematyczne tworzenie krążących wariantów SARS-Cov-2
„W ciągu ostatnich trzech lat wirus ciężkiego ostrego zespołu oddechowego (SARS-CoV-2) wielokrotnie wywoływał pandemie, generując różne zmutowane warianty, od wariantu Alfa po Omicron. W tym badaniu chcieliśmy wyjaśnić procesy ewolucyjne prowadzące do powstania wariantów Omicron SARS-CoV-2, koncentrując się na wariantach Omicron z wieloma mutacjami aminokwasowymi w białku kolca wśród izolatów SARS-CoV-2. Aby określić kolejność mutacji prowadzących do powstania wariantów SARS-CoV-2 Omicron, porównaliśmy sekwencje 129 izolatów związanych z Omicron BA.1, 141 związanych z BA.1.1 i 122 związanych z BA.2 oraz podjęto próbę wyjaśnienia procesów ewolucyjnych wariantów SARS-CoV-2 Omicron, w tym kolejności mutacji prowadzących do ich powstania oraz wystąpienia rekombinacji homologicznej. W rezultacie doszliśmy do wniosku, że powstanie części izolatów Omicron BA.1, BA.1.1 i BA.2 nie było produktem ewolucji genomu, jak to jest powszechnie obserwowane w przyrodzie, takiej jak akumulacja mutacji i homologicznych rekombinacje. Co więcej, badanie 35 rekombinowanych izolatów wariantów Omicron BA.1 i BA.2 potwierdziło, że warianty Omicron były obecne już w 2020 r. Analiza wykazała, że warianty Omicron powstały w wyniku zupełnie nowego mechanizmu, którego nie da się wytłumaczyć wcześniejszą biologią, oraz wiedza o tym, jak powstały warianty SARS-CoV-2 skłania do ponownego rozważenia pandemii SARS-CoV-2
(…)
Celem tego badania jest wskazanie, że SARS-CoV-2 przeszedł niewyobrażalne mutacje w oparciu o konwencjonalne mechanizmy mutacji koronawirusa i mamy nadzieję, że możliwość sztucznego stworzenia zostanie uwzględniona w poważnych dyskusjach na temat powstawania wariantów SARS-CoV-2.” – Źródło: Raw data for „Unnatural evolutionary processes of SARS-CoV-2 variants and possibility of deliberate natural selection”
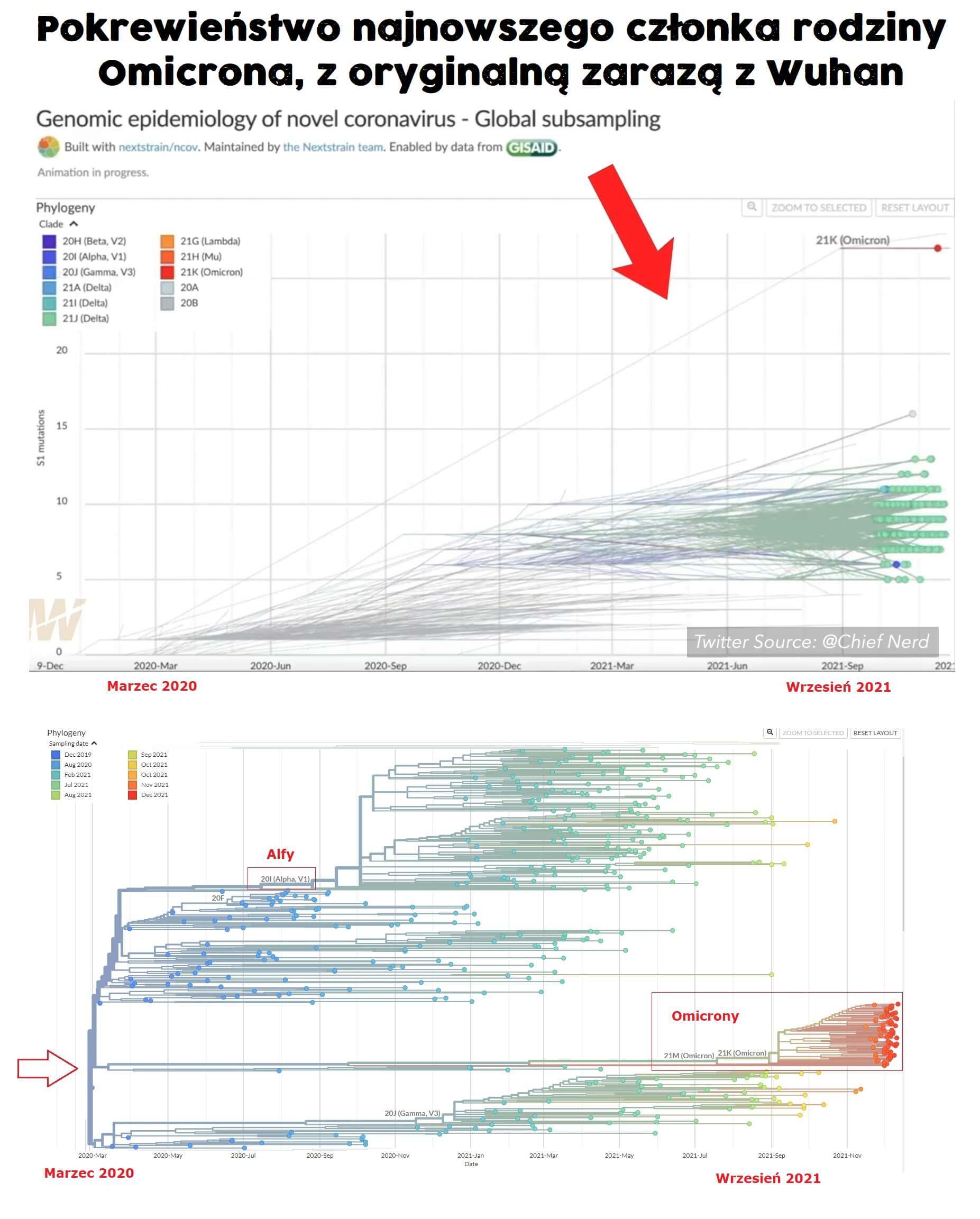
5 sierpnia 2023 roku japoński zespół badawczy opublikował przeddruk [preprint] pod tytułem „Unnaturalness in the evolution process of the SARS-CoV-2 variants and the possibility of deliberate natural selection [Nienaturalność w procesie ewolucji wariantów SARS-CoV-2 a możliwość celowej selekcji naturalnej]”, który wydaje się zawierać najważniejsze i najbardziej szokujące rewelacje dla ery COVID-19.
Atsuki Tanaka i Takayuki Miyazawa z Uniwersytetu Medycznego w Osace i Uniwersytetu w Kioto chcieli prześledzić historyczną ewo lucję wariantu omicron wirusa SARS-CoV2, badając sekwencje wirusa znalezione „na wolności” i zdeponowane w publicznych bazach danych.
W ten sposób znaleźli około 100 oddzielnych podwariantów omicrona, które nie mogły powstać w wyniku naturalnych procesów. Istnienie tych wariantów wydaje się stanowić ostateczny dowód na tworzenie i uwalnianie wirusów z podgrupy SARS-Cov-2 odpowiedzialnych za Covid-19 na dużą skalę w laboratoriach.
Co więcej, warianty te wydają się tworzyć kompleksowe panele mutacji typowych dla tych stosowanych w eksperymentach typu „odwróconej genetyki [reverse genetics]” w celu systematycznego testowania właściwości różnych części wirusów.
Autorzy znaleźli również dokładne dopasowanie do wariantów omicron w sekwencjach pochodzących z Puerto Rico, które zostały zdeponowane w bazach danych w 2020 roku – ponad rok przed ogłoszeniem odkrycia omicron w RPA.
W połączeniu z obserwacjami niewiarygodnie niskiej liczby „cichych” mutacji w wariantach SARS-CoV2, Tanaka i Miyazawa twierdzą, że wszystkie warianty pojawiające się od pierwotnego wybuchu epidemii w Wuhan są nienaturalne i spekulują, że reprezentują one program eksperymentalny do testowania determinantów zakaźności i patogenności SARS-CoV2 w populacji globalnej.
[Mutacje niesynonimiczne (zwane również jako cicha substytucja lub podstawienie) zmieniają sekwencje białek i często podlegają selekcji naturalnej. Mutacje synonimiczne są intuicyjnie uważane za funkcjonalnie ciche i ewolucyjnie neutralne, które nie mają zauważalnego wpływu na fenotyp organizmu. Wyrażenie cicha mutacja jest często używane zamiennie z wyrażeniem synonimiczna mutacja; jednakże mutacje synonimiczne nie zawsze są ciche i odwrotnie.
Synonimiczne podstawienia zasad (tj. takie, które nie powodują zmian aminokwasów) występują prawie zawsze ze znacznie większą szybkością niż podstawienia [mutacje] niesynonimiczne.]
DODATEK: widoczne „panele” mutacji rewersyjnych można wyjaśnić, jeśli SARS-CoV2 istnieje jako „quasigatunek wirusa” – to znaczy
„struktura populacji składająca się z niezwykle dużej liczby wariantów genomów, określanych jako spektrum mutantów, rojami zmutowanych lub chmurami zmutowanych.Napędzane wysokim współczynnikiem mutacji, mutanty pojawiają się stale i zmieniają swoją względną częstotliwość w miarę postępu replikacji wirusa. Termin quasigatunek został zaczerpnięty z teorii pochodzenia życia, w której prymitywne replikony składały się z zmutowanych rozkładów, co stwierdzono eksperymentalnie z obecnymi wirusami RNA.” – PLoS Genet. 2019 Oct 17;15(10):e1008271; Viral quasispecies
Nie wyjaśniałoby to jednak braku cichych mutacji w wariancie omicron i innych wariantach, ani wykrycia sekwencji omicron w 2020 roku w Puerto Rico. Podziękowania dla Josha Mitteldorfa za zwrócenie na to uwagi.
Tło: naturalna ewolucja przebiega poprzez akumulację mutacji
Przed opisaniem badania i jego wyników warto szybko przejrzeć podstawowe zasady ewolucji wirusów (i wszystkich form życia). Jeśli je znasz, możesz to pominąć.
SARS-CoV2, podobnie jak wszystkie wirusy i organizmy, jest definiowany przez swoją informację genetyczną – którą można traktować jako ciąg lub sekwencję liter. W większości organizmów ciąg ten składa się z DNA, ale SARS-CoV2 i niektóre inne wirusy wykorzystują ciągi RNA, blisko spokrewnionej cząsteczki, aby zapewnić tę funkcję przechowywania informacji.
Materiał genetyczny (DNA lub RNA) jest podzielony na „geny” – odcinki sekwencji genetycznej, z których każdy koduje białko. Białka są aktywnymi cząsteczkami, które są syntetyzowane przez komórkę, wykorzystując gen jako wzorzec. Białka są również sekwencjami, ale mają bardzo różne bloki budulcowe. Nie istnieją jako proste ciągi, które tylko przechowują informacje, ale zamiast tego tworzą złożone struktury trójwymiarowe [3D], które cechują się aktywnością biologiczną.
Kiedy organizm się rozmnaża, sekwencja genetyczna (DNA lub RNA) jest kopiowana i przekazywana następnemu pokoleniu. Mechanizm kopiowania jest jednak podatny na błędy i czasami dochodzi do zmiany lub „mutacji”. Kolejne pokolenia będą przekazywać tę sekwencję, więc mutacje naturalnie gromadzą się w czasie.
Poniższy diagram ilustruje, jak to się dzieje – oryginalna sekwencja DNA gromadzi 4 kolejne mutacje.
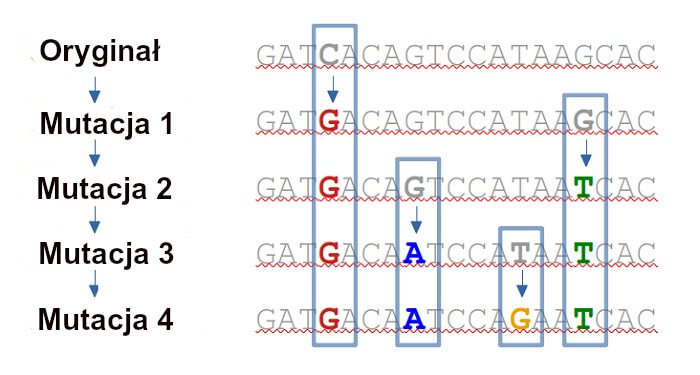
Wpływ mutacji na organizm decyduje o tym, czy utrzymują [przeżyją] się one w przyrodzie
Warto wziąć pod uwagę, że istnieje kilka różnych ogólnych rodzajów mutacji, ocenianych na podstawie ich wpływu na organizm:
— Wiele z nich nie będzie miało żadnego wpływu. Niektóre indywidualne zmiany w literze DNA/RNA nie zmieniają sekwencji kodowanego białka. Takie „synonimiczne” mutacje zwykle nie mają żadnego wpływu, więc po prostu gromadzą się z czasem w naturalnej ewolucji. Jak zobaczymy, brak mutacji synonimicznych jest ważnym znakiem, że ewolucja omikronów i innych wariantów nie jest naturalna.
— Spośród tych, które mają wpływ, zdecydowana większość będzie szkodliwa – z tego samego powodu, dla którego losowa zmiana części dowolnego funkcjonującego systemu może go zepsuć. Takie mutacje szybko znikną z populacji.
— Bardzo rzadko mutacja spowoduje zmianę z korzystnym efektem. Mutacje te będą następnie rozprzestrzeniać się w przyszłych pokoleniach – ponieważ organizmy je niosące przetrwają i będą rozmnażać się bardziej efektywnie.
Tak więc wszystkie formy życia stopniowo gromadzą mutacje – niektóre ciche, a niektóre korzystne. Na tym polega ewolucja.
Większość losowych pojedynczych mutacji faktycznie zmieni kodowany aminokwas. Ale przytłaczająca większość z nich po prostu zepsuje białko, więc nadal prawdą jest stwierdzenie, że synonimiczne ciche mutacje powinny początkowo przewyższać liczbę mutacji funkcjonalnych.
„Nienaturalna ewolucja” omikronów
Rozważmy teraz konkretny przypadek wariantu omicron wirusa SARS-CoV2 i jego (przypuszczalnej) ewolucji z oryginalnego szczepu Wuhan.
W tym artykule będziemy podążać za Tanaką i Miyazawą, koncentrując się tylko na jednej sekcji ciągu informacji genetycznej wirusa – genie kodującym osławione białko kolcowe. Rozważają oni trzy oficjalnie uznane warianty omicron – BA1, BA1.1 i BA2. Na razie przyjrzymy się tylko BA1.
Podobnie jak w przypadku całej ewolucji, zmiany w białku kolcowe zachodzą poprzez stopniową akumulację mutacji w sekwencji genetycznej, która je koduje. W przypadku wariantu omicron BA1 istnieje 37 mutacji niesynonimicznych – to znaczy punktów, w których sekwencja wytwarzanego białka kolca różni się od sekwencji oryginalnego wariantu Wuhan.
Tanaka i Miyazawa chcieli wykorzystać publiczne bazy danych, w których badacze z całego świata deponują znalezione przez siebie sekwencje wirusowe, aby prześledzić historię ewolucji białka kolcowego wariantu omicron BA1 – czyli odpowiedzieć na pytanie: w jakiej kolejności nagromadziło się te 37 mutacji?
Śledzenie kolejności akumulacji mutacji omikronowych
Są na to dwa oczywiste sposoby – praca do przodu lub do tyłu.
W podejściu do przodu można szukać w bazach danych wersji sekwencji, które mają tylko jedną z 37 mutacji omikronowych, ale poza tym są identyczne z oryginalnym szczepem. Ta samotna mutacja musiała być pierwszą. Następnie można powtórzyć proces, aby zidentyfikować drugą, trzecią itd.
Jednak warianty niosące bardzo wczesne mutacje byłyby rzadkie w globalnej populacji SARS-CoV2 i mogą nie pojawić się w bazach danych.
Pewniejszym podejściem, przyjętym przez Tanakę i Miyazawę, jest praca wstecz. Oznacza to rozpoczęcie od ustalenia, która z 37 mutacji jest ostatnią lub najnowszą.
Aby to zrobić, musisz znaleźć sekwencję, która zawiera wszystkie mutacje z wyjątkiem jednej – i musi to być najnowsza mutacja.
Tak więc Tanaka i Miyazawa wykonali serię 37 zapytań do bazy danych przy użyciu sekwencji, z których każda nie zawierała tylko jednej z mutacji omicron BA1 – rozumując, że jedna z 37 powinna znaleźć dopasowanie, wskazując ostatnią mutację w ewolucyjnym postępie do BA1.
Ciekawie jest wyobrazić sobie siebie w sytuacji tych badaczy, uruchamiających zapytania dla każdej z 37 mutacji, być może zastanawiających się, która z nich okaże się najnowsza, i otrzymujących odpowiedź… …WSZYSTKIE*.
Cóż, wszystkie oprócz jednej – co nie robi żadnej istotnej różnicy.
Ich mózgi musiały eksplodować.
Panel wariantów z indywidualnie odwróconymi mutacjami omicron BA1 nie może powstać naturalnie
Na poniższej rycinie (ryc. 2A z artykułu) każdy wiersz reprezentuje wariant omicron BA1 znaleziony „na wolności”.
Kolumny reprezentują każdą z mutacji omicron. Jeśli komórka jest kolorowa, oznacza to, że wariant zawiera mutację. Białe komórki wskazują na brak mutacji, a sekwencja białka kolca jest w tym momencie identyczna z oryginalnym szczepem Wuhan.
Jeśli uważasz, że tabela wygląda strasznie schludnie, masz rację. Pokazuje ona, że dla wszystkich mutacji z wyjątkiem jednej w podwariancie omikronowym BA1, istnieje szczep, w którym ta mutacja – sama w sobie – jest nieobecna.
W naturalnej ewolucji przez skumulowaną mutację, każdy wariant ma tylko jednego rodzica – ponieważ powstał w wyniku pojedynczej mutacji. Tak więc, biorąc pod uwagę wartość nominalną, wyniki te sugerują, że jeden z wariantów jest rodzicem omicron BA1 (nie możemy powiedzieć, który), a wszystkie pozostałe są dziećmi.
Możemy teraz odpowiedzieć na pierwotne pytanie Tanaki i Miyazawy i określić naturalną historię ewolucji wariantu omikronu BA1 sugerowaną przez te wyniki:
1.Oficjalnie uznany szczep BA1 powstaje, gdy wystąpi ostatnia z 37 mutacji (nie wiemy, która to jest);
2.Następnie BA1 przechodzi 35 oddzielnych, równoległych zmian, z których każda doskonale odwraca jedną z tych mutacji do sekwencji oryginalnego szczepu Wuhan.
To absurd. Doskonała rewersja mutacji na taką skalę jest całkowicie nieprawdopodobna w jakimkolwiek naturalnym procesie.
Warianty znalezione przez Tanakę i Miyazamę można najlepiej opisać jako „panel” mutacji rewersyjnych. Tego rodzaju panel jest dokładnie tym, co badacz stworzyłby, aby systematycznie testować wpływ różnych elementów wirusa na jego aktywność.
Kompleksowe „panele” rewersji występują również w przypadku innych oficjalnych wariantów omicron
Naukowcy przyjrzeli się również dwóm innym uznanym wariantom omicron w szerokim obiegu: BA1.1 i BA2. Co ciekawe, znaleźli te same „panele” mutacji rewersyjnych dla obu z nich.
BA1.1 jest bardzo podobny do BA1 – ma tylko jedną dodatkową mutację, w porównaniu do szczepu Wuhan, w sumie 38.
Kiedy Tanaka i Miyazawa przeprowadzili kwerendę, pomijając każdą z tych mutacji z osobna, znaleźli 37 z 38 istniejących „na wolności”.
Wariant BA2 różni się dość znacznie od BA1 – mają 14 wspólnych mutacji w odniesieniu do szczepu Wuhan, ale BA2 ma tylko 17 dalszych (różnych) mutacji, co daje łącznie 31.
Kiedy szukano wariantów pozbawionych tych indywidualnych mutacji, znaleziono 29 z 31 „na wolności”.
„Rekombinacja” lub zamiana materiału genetycznego między różnymi wirusami również nie może wyjaśnić wariantów Omicron
Powyższa dyskusja odnosi się do ewolucji poprzez progresywną akumulację mutacji, w której każdy nowy wariant jest wytwarzany przez mutację pojedynczego rodzica.
Istnieje inny mechanizm, dzięki któremu wirusy i inne formy życia mogą ewoluować. „Rekombinacja” polega na zamianie fragmentów materiału genetycznego między dwoma różnymi wariantami. Czy zaobserwowane warianty mogły powstać w wyniku zamiany materiału genetycznego między omicron BA1 a oryginalnym wirusem Wuhan?
Tanaka i Miyazawa dokładają wszelkich starań, aby rozważyć taką możliwość, ale z łatwością ją wykluczają.
Po pierwsze, rekombinacja wymagałaby, aby wirusy omicron BA1 i inne wirusy przodków były obecne w tej samej komórce w tym samym czasie – ponieważ rekombinacja może wystąpić tylko w komórce, podczas fazy replikacji wirusa. Będzie to niezwykle rzadkie, biorąc pod uwagę zaangażowane częstotliwości i wymóg stworzenia tak wielu mutacji rewersyjnych – szczególnie biorąc pod uwagę czas fal różnych wariantów, jak omówiono w artykule.
Wyjaśnienie tych rewersji przez rekombinację oznaczałoby, że odcinek RNA w omikronie BA1 zawierający mutację, która ma zostać odwrócona, musiałby zostać całkowicie zamieniony, tak aby mutacje po obu stronach pozostały nienaruszone. Musiałyby istnieć dwa „skrzyżowania” między wariantami, po jednym z każdej strony mutacji. Jednak krzyżowanie wymaga wyrównania odcinka wspólnej sekwencji między dwoma szczepami. W przypadku niektórych mutacji przerwa po obu stronach następnej mutacji jest po prostu niewystarczająco duża, aby pomieścić te krzyżowania, w związku z czym rekombinacja jest niemożliwa.
Rekombinacja pozostawiłaby również ślady w regionach flankujących wirusa po obu stronach genu białka kolca – a tych nie znaleziono.
Warianty omicron w próbkach z Puerto Rico – ponad rok przed oficjalnym wykryciem omicronów
Tanaka i Miyazawa byli więc w stanie wykazać, że rekombinacja nie może wyjaśnić panelu mutacji rewersyjnych, które znaleźli. Ale rozważając tę możliwość, natknęli się na jeszcze więcej dowodów, które rodzą fundamentalne pytania dotyczące historii omicronów.
Gdy przeprowadzili kwerendy w bazie danych w poszukiwaniu oznak, że rekombinacja była zaangażowana, znaleźli dopasowania do sekwencji z Puerto Rico, która została przesłana w 2020 roku. Dalsze wyszukiwania znalazły 29 wariantów przypisanych Puerto Rico, które dokładnie pasują do omicron BA1 lub BA2, w oparciu o sekwencje białek kolcowych.
Wszystkie te sekwencje zostały zdeponowane w bazie danych w 2020 roku, ponad rok przed ogłoszeniem wykrycia omicron w RPA w listopadzie 2021 roku.
Brak mutacji „synonimicznych” silnie sugeruje sztuczne pochodzenie wariantów SARS-CoV2
Jakby tego było mało, Tanaka i Miyazawa wskazują na kolejną linię dowodów, która sama w sobie jest prawdopodobnie wystarczająca, aby stwierdzić, że warianty omicron są nienaturalne.
Powyższe wyjaśnienie mutacji wskazuje, że w naturze można spodziewać się:
— niektórych, które mają istotny i korzystny wpływ na organizm; oraz
— niektórych „cichych” lub „synonimicznych”, które nie wpływają na białka wytwarzane z RNA/DNA i nie powinny zmieniać zdolności organizmu do rozmnażania się.
Te „synonimiczne” mutacje, które w rzeczywistości nie zmieniają odpowiedniego białka, są, jak można się spodziewać, początkowo znacznie częstsze niż te, które wpływają na samo białko. Zwykle nie mają one wpływu na zdolność wirusa do przetrwania, więc po prostu gromadzą się naturalnie w czasie, wraz z bardziej funkcjonalnym nabywaniem rzadszych korzystnych mutacji niesynonimicznych.
Jednak wspomniane tutaj oficjalne warianty omicron mają tylko jedną mutację synonimiczną w genie kodującym białko kolcowe – w porównaniu do od 31 do 38 mutacji niesynonimicznych.
To nie ma sensu. Naturalna ewolucja zawsze powinna tworzyć ciche mutacje synonimiczne w większym tempie niż mutacje niesynonimiczne, które mogą przetrwać tylko wtedy, gdy wbrew wysokim szansom losowo powodują ulepszenie projektu białka, które kodują.
Autorzy wskazują, że ta nieprawdopodobna obserwacja nie ogranicza się do omikronów:
„Jeśli chodzi o zmienność genetyczną białka S tych wariantów, większość mutacji nie była synonimiczna (ryc. 1). Nie było mutacji synonimicznych w wariantach Alfa, Beta, Gamma, Delta lub Mu, [i] tylko po jednej w wariantach Lambda i Omicron.”
Panele rewersji Omicron wydają się być częścią systematycznego eksperymentu
Obecność „na wolności” prawie kompletnych paneli doskonałych indywidualnych rewersji praktycznie każdej mutacji w 3 oddzielnych liniach omicronów nie może być naturalna. Zamiast tego wygląda to dokładnie jak systematyczne ćwiczenie z „genetyki odwrotnej” w celu przetestowania wpływu każdej mutacji omicron na zachowanie wirusa.
Oczywiste jest, że niektóre lub wszystkie warianty omikronów zostały zsyntetyzowane w laboratorium, z którego zostały w jakiś sposób uwolnione, jako część celowego programu. W połączeniu z brakiem mutacji synonimicznych w innych wariantach sugeruje to, że wszystkie warianty opisane po oryginalnym szczepie Wuhan mają sztuczne pochodzenie.
Autorzy sugerują, że znalezione przez nich warianty są rzeczywiście częścią eksperymentu mającego na celu scharakteryzowanie białka kolcowego i wpływu mutacji na zachowanie wirusa:
„Rzeczywiście, brak dotychczasowych ustaleń, że wiele różnych zaobserwowanych mutacji, zwłaszcza we wczesnych wariantach, jest rzeczywiście związanych ze zwiększoną infekcją wirusową (van Dorp i in., 2020), potwierdza hipotezę, że każdy wariant został sztucznie zsyntetyzowany w celu zidentyfikowania aminokwasów białka S odpowiedzialnych za zakaźność i patogenność.”
Wniosek: to zmienia wszystko
Jeśli obserwacje i wnioski zawarte w tym artykule są poprawne – a z wyjątkiem czystego oszustwa, obejmującego fałszywe depozyty w bazach danych sekwencji, z pewnością wydają się być – to dostarczają niepodważalnych dowodów na to, że cała historia SARS-CoV2, przynajmniej po pojawieniu się oryginalnego szczepu, jest sztuczna.
Ktoś, gdzieś, naprawdę robi to wszystko celowo.
Zobacz na: Jak się tworzy konsensus „naukowy” – dr Anthony Fauci i dr Francis Collins
Laureat Nagrody Nobla Luc Montagnier twierdzi, że COVID-19 pochodzi z laboratorium
Li-Meng Yan o laboratoryjnej genezie koronawirusa
Dr Dolores Cahill: Jak SARS-CoV-2 zmodyfikowano w laboratorium?
SARS-CoV-2 w kontekście patentów sprzed 20 lat – dr David Martin
Anthony Fauci kłamał, a ludzie umierali – dr Chris Martenson
Anthony Fauci: „Korzyści ze zgromadzonych danych przeważają nad ryzykiem związanym z pandemią” – dr Chris Martenson
USA: Dr Deborah Birx chwali samą siebie, jednocześnie ujawniając ignorancję, zdradę i oszustwo