Aktywacja komórek glejowych i stan zapalny układu nerwowego w mózgu pacjentów z autyzmem – Dr Diana L. Vargas,1,2 dr Caterina Nascimbene,1-3 magister nauk o zdrowiu Chitra Krishnan,1 dr Andrew W. Zimmerman1,4 i dr Carlos A. Pardo1,2,5
Źródło: Neuroglial Activation and Neuroinflammation in the Brain of Patientswith Autism – Vargas – Ann Neurol. 2005 Jan;57(1):67-81.
https://www.ncbi.nlm.nih.gov/pubmed/15546155

Dr Diana Lucia Vargas Vives
1Katedra Neurologii i 2Wydział Neuroimmunologii i Chorób Zakaźnych, Szkoła Medyczna Uniwersytetu Johnsa Hopkinsa, Baltimore, Maryland, 3Wydział Neurologii Uniwersytetu w Mediolanie, 4Kennedy Krieger Institute i 5Wydział Patologii Szkoły Medycznej Uniwersytetu Johnsa Hopkinsa w Baltimore, Maryland.
Artykuł złożony do publikacji 8 lipca 2004 roku, w zmienionej formie 14 sierpnia. Przyjęty do publikacji 14 sierpnia 2004 r.
Opublikowany w Internecie 15 listopada 2004 r. w Wiley InterScience
(www.interscience.wiley.com). DOI: 10.1002/ana.20315
Adres korespondencyjny dra. Pardo na Wydziale Neurologii Szkoły Medycznej Uniwersytetu Johnsa Hopkinsa, Pathology 627, 600 North Wolfe Street, Baltimore, MD 21287; E-mail: pa***@**mi.edu
Abstrakt
Autyzm to zaburzenie neurorozwojowe charakteryzujące się upośledzonymi zdolnościami komunikacyjnymi oraz interakcji społecznej, przy czym może mu towarzyszyć upośledzenie umysłowe i epilepsja. Jego przyczyna pozostaje nieznana, mimo dowodów że czynniki genetyczne, środowiskowe oraz immunologiczne mogą odgrywać rolę w jego patogenezie. W celu ustalenia czy mechanizmy o podłożu immunologicznym uczestniczą w patogenezie autyzmu, zastosowaliśmy metodę immunocytochemii, testy cytokin, a także test ELISA dla zbadania tkanek mózgu i płynu mózgowo-rdzeniowego (ang. cerebrospinal fluid, CSF) pacjentów autystycznych i określiliśmy skalę reakcji komórek glejowych i zapalnych oraz profile ekspresji ich cytokin. Do badań morfologicznych użyto tkanek mózgu z móżdżku, zakrętu czołowego środkowego i zakrętu obręczy uzyskanych podczas autopsji od 11 pacjentów z autyzmem. Do określenia profili cytokin użyto świeżo mrożonych tkanek pobranych od siedmiu pacjentów, zaś CSF od sześciu żywych pacjentów autystycznych. Przedstawiamy aktywny proces zapalny w układzie nerwowym w korze mózgowej oraz istocie białej, a zwłaszcza w móżdżku pacjentów z autyzmem. Badania immunocytochemiczne wykazały wyraźną aktywację mikrogleju i astrogleju, zaś profile cytokin uwidoczniły, że białko chemotaktyczne dla makrofagów typu 1 (MCP-1) i nowotworowy czynnik wzrostu Beta1 [β1], pochodzące z komórek glejowych, były najbardziej powszechnymi cytokinami w tkance mózgu. CFS wykazał unikalny profil prozapalny cytokin, w tym wyraźny wzrost poziomu MCP-1. Nasze ustalenia wskazują, że wrodzone reakcje neuroimmunologiczne odkrywają patogeniczną rolę u nieokreślonej części pacjentów autystycznych, co z kolei sugeruje, iż w przyszłości metody leczenia powinny być może uwzględniać modyfikowanie odpowiedzi komórek glejowych w mózgu.
Autyzm to powszechne zaburzenie neurorozwojowe charakteryzujące się upośledzeniem funkcji społecznych, behawioralnych i komunikacyjnych.[1,2] Objawy występują przed ukończeniem 36 miesiąca życia, zaś regres lub utrata umiejętności występują u 30% dzieci dotkniętych chorobą, zwykle w wieku od 18 do 24 miesięcy. Zespół ten jest klinicznie heterogeniczny i można go powiązać z częstością do 10% pacjentów z dobrze opisanymi zaburzeniami neurologicznymi i genetycznymi, takimi jak stwardnienie guzowate oraz zespoły łamliwego chromosomu X, Retta i Downa, choć u większości pacjentów przyczyny są nadal nieznane.[3,4] Najnowsze badania epidemiologiczne wskazują, że liczba przypadków zaburzeń ze spektrum autyzmu wzrosła w ostatnich latach z 1 na 500 do 1 na 250 dzieci, być może na skutek postępu w metodach diagnostycznych.[5-7]
Pomimo że wciąż niedostatecznie rozumiemy podstawy neurobiologiczne autyzmu, kilka linii badań wspiera obecnie pogląd, że do jego rozwoju przyczyniają się czynniki genetyczne, środowiskowe, neurologiczne oraz immunologiczne.[3,48-10] Badania neuropatologiczne wykazują, że anomalia w budowie cytoarchitektonicznej kory mózgowej i struktur podkorowych, a także zmniejszona liczba komórek Purkiniego w móżdżku to zjawiska występujące najbardziej konsekwentnie w tkance mózgu pobranej pośmiertnie od pacjentów z autyzmem. Wskazują, że defekty w dojrzewaniu neuronów i strukturze kory mózgowej mogą odpowiadać za niektóre problemy neurologiczne obserwowane u chorych na autyzm.[11-13]
Wskazywano, że potencjalnym mechanizmem patogenezy autyzmu jest nieprawidłowe funkcjonowanie układu odpornościowego.[14] Kilka badań krwi obwodowej wykazało rozmaite anomalia, jak dysfunkcja limfocytów T, produkcja autoprzeciwciał i zwiększona liczba cytokin prozapalnych.[9,15-18] Zwracano również uwagę na potencjalną rolę matczynych przeciwciał jako czynnika patogenicznego.[19] Badania płynu mózgowo-rdzeniowego (CSF), takie jak określenie standardowej liczby komórek, elektroforeza białek lub pomiary kwasu chinolinowego i neopteryny, nie wykazały żadnego dowodu na wystąpienie stanu zapalnego.[20] Pomimo rosnącego zainteresowania prawdopodobnymi mechanizmami immunologicznymi patogenezy autyzmu, nie istnieje bezpośredni dowód łączący odkrycia we krwi obwodowej z aktywnością immunologiczną w mózgu pacjentów autystycznych.[21] Badania neuropatologiczne w niewielkim stopniu koncentrowały się na aktywności immunologicznej czy aktywności komórek glejowych u chorych na autyzm, zaś najbardziej kompleksowe badanie pośmiertne nie wykazało żadnych zmian zapalnych ani reakcji astrogleju.[11] W opisach zaledwie kilku badań znalazła się wzmianka o glejozie i zmianach zapalnych.[12,22] Taki stan neurozapalny, jeśli występuje w mózgu, może zarówno uczestniczyć w i być skutkiem nieprawidłowego rozwoju i funkcjonowania ośrodkowego układu nerwowego (ang. central nervous system, CNS) u osób z autyzmem. Aby zbadać czy mechanizmy o podłożu immunologicznym uczestniczą w patogenezie autyzmu w odniesieniu do ośrodkowego układu nerwowego, zbadaliśmy tkanki mózgu oraz płyn mózgowo-rdzeniowy [CSF] pacjentów autystycznych i określiliśmy skalę reakcji komórek glejowych oraz ich profile ekspresji cytokin.
Stan zapalny układu nerwowego w mózgu pacjentów z autyzmem
Materiały i metody
Informacje dotyczące pacjentów
Tkanki mózgu pacjentów autystycznych i nieneurologicznych w grupie kontrolnej zostały uzyskane z Autism Tissue Program [ATP] z banków mózgów uniwersytetu Harvard, Uniwersytetu w Miami i Uniwersytetu w Maryland. Wszystkie przypadki autyzmu spełniają kryteria ustalone w Diagnostic and Statistical Manual-IV i zatwierdzone w wywiadzie ADI-R (ang. Autism Diagnostic Interview-Revised).[23,24] ADI-R był wcześniej stosowany przez naukowców z Programu Tkanek Autystycznych [ATP] jako kryterium włączenia do repozytorium. Dodatkowe informacje kliniczne i neurologiczne także pochodzą z ATP. Charakterystyka demograficzna wszystkich pacjentów autystycznych oraz osób w grupie kontrolnej objętych badaniem została przedstawiona w Tabeli 1. Informacje dotyczące historii epilepsji, upośledzenia umysłowego i regresji u pacjentów z autyzmem również znajdują się w Tabelach 1 i 2.
Tabela 1. Informacje dotyczące tkanki mózgu pacjenta
| Nr przypadkua | Diagnoza | Wiekb (lata) | Płeć | Czas od zgonu Godziny | Przyczyna zgonu | Epilepsja | Regresja | Upośledzenie umysłowe |
| 1349c | Autyzm | 5 | M | 39 | Utonięcie | Nie | Nie | Tak |
| 1174c | Autyzm | 7 | F | 14 | Nagły zgon | Tak | Nie | Tak |
| B5013d | Autyzm | 7 | M | 40 | Utonięcie | Nie | Nie | Tak |
| 2004d | Autyzm | 8 | M | 23 | Utonięcie | Nie | Nie | Tak |
| 1182c | Autyzm | 9 | F | 24 | Niewydolność oddechowa | Tak | Tak | Tak |
| 797e | Autyzm | 9 | M | 13 | Utonięcie | Nie | Brak danych | Brak danych |
| B4925e | Autyzm | 9 | M | 27 | Nagły zgon | Tak | Nie | Tak |
| 3714d | Autyzm | 10 | M | 30 | Utonięcie | Nie | Brak danych | Tak |
| B4323d | Autyzm | 14 | M | 10 | Hipertermia | Nie | Nie | Nie |
| 1638c | Autyzm | 20 | F | 50 | Nagły zgon | Tak | Tak | Tak |
| B5144c | Autyzm | 20 | M | 23 | Obrażenia | Nie | Tak | Tak |
| 3711d | Autyzm | 25 | M | 26 | Znalaziony martwy, nieznana | Tak | Brak danych | Brak danych |
| 3663d | Autyzm | 27 | M | 30 | Zespół neuroleptyczny | Tak | Nie | Tak |
| 2802d | Autyzm | 29 | M | 24 | Zachłyśnięcie | Nie | Brak danych | Tak |
| B4541d | Autyzm | 44 | M | 30 | Ostry zawał mięśnia sercowego | Nie | Nie | Tak |
| 1377c | Grupa kontrolna | 5 | F | 20 | Utonięcie | Nie | Nie | |
| 1706c | Grupa kontrolna | 8 | F | 20 | Odrzucenie przeszczepu | Nie | Nie | |
| 1860c | Grupa kontrolna | 8 | M | 5 | Nagły zgon, arytmia serca | Nie | Nie | |
| 629d | Grupa kontrolna | 7 | M | 18 | Przypadkowy zgon | Nie | Nie | |
| 1407c | Grupa kontrolna | 9 | F | 20 | Astma | Nie | Nie | |
| 2149d | Grupa kontrolna | 16 | M | 13 | Rana postrzałowa | Nie | Nie | |
| 1862c | Grupa kontrolna | 20 | M | 6 | Obrażenia | Nie | Nie | |
| 3706d | Grupa kontrolna | 27 | M | 21 | Powieszenie | Nie | Nie | |
| 3231d | Grupa kontrolna | 37 | M | 24 | Uduszenia | Nie | Nie | |
| 2845d | Grupa kontrolna | 37 | M | 21 | Choroba serca | Nie | Nie | |
| B3706c | Grupa kontrolna | 40 | M | 28 | Obrażenia | Nie | Nie | |
| B4192e | Grupa kontrolna | 46 | M | 25 | Nagły zgon, nieznana | Nie | Nie |
a identyfikator Autism Tissue Program (ATP).
b Średni wiek w grupie autyzmu: 16 lat; średnia liczba godzin po zgonie: 18.
c Przypadki, w których jedynie mrożone tkanki były dostępne do badań za pomocą testów cytokin.
d Przypadki, w których jedynie utrwalone tkanki były dostępne do badań morfologicznych.
e Przypadki, w których mrożone i utrwalone tkanki były dostępne do badań morfologicznych i badań z użyciem testów cytokin.
Tabela 2. Informacje dotyczące CSF pacjentów.
| Nr przypadku | Wiek (lata) | Płeć | Grupa | Diagnoza | Regresja | Upośledzenie umysłowe |
| 5 | 4 | F | Autyzm | Autyzm | Tak | Tak |
| 6 | 4 | M | Autyzm | Autyzm | Tak | Nie |
| 8 | 6 | M | Autyzm | Autyzm | Tak | Tak |
| 9 | 10 | M | Autyzm | Autyzm | Tak | Tak |
| 11 | 6 | F | Autyzm | Autyzm | Tak | Tak |
| 12 | 3 | M | Autyzm | Autyzm | Tak | Nie |
| 5061 | 36 | F | Grupa kontrolna | Bóle głowy | Nie dotyczy | Nie |
| 3685 | 26 | F | Grupa kontrolna | Bóle głowy | Nie dotyczy | Nie |
| 7108 | 42 | M | Grupa kontrolna | Depresja | Nie dotyczy | Nie |
| 7384 | 45 | M | Grupa kontrolna | Ostry zespół mózgowy | Brak danych | Nie |
| 150 | 35 | F | Grupa kontrolna | Rzekomy guz mózgu | Nie dotyczy | Nie |
| 400 | 26 | F | Grupa kontrolna | Rzekomy guz mózgu | Nie dotyczy | Nie |
| 500 | 12 | M | Grupa kontrolna | Rzekomy guz mózgu | Nie dotyczy | Nie |
a Numer identyfikacyjny w repozytorium CSF w Kennedy Krieger Institute w Baltimore, Maryland lub w Repozytorium CSF Wydziału Neurologii Uniwersytetu Johnsa Hopkinsa w Baltimore.
CSF = płyn mózgowo-rdzeniowy
Upośledzenie umysłowe zostało zdefiniowane jako pełne IQ pomniejszone o 70, któremu towarzyszą osłabione funkcje poznawcze; regresję zdefiniowano jako utratę nabytych wcześniej umiejętności językowych i społecznych, przy czym utrata obu tych rodzajów umiejętności nastąpiła we wczesnym dzieciństwie.
Przetwarzanie tkanki mózgu
Utrwalone i mrożone próbki tkanki mózgu pochodziły ze stowarzyszonych w ATP banków mózgów (patrz Tabela 1). Utrwalone tkanki mózgu z zakrętu czołowego środkowego (ang. middle frontal gyrus, MFG), przedniego zakrętu obręczy (ang. anterior cingulate gyrus, ACG) i z półkuli móżdżku (ang. cerebellar hemisphere, CBL) zostały wyselekcjonowane spośród mózgów pobranych w czasie autopsji od pacjentów autystycznych (n = 11) oraz pacjentów z grupy kontrolnej (n = 6) (patrz Tabela 1). Tylko 3 z 11 mózgów pacjentów z autyzmem posiadały świeżo mrożone tkanki dostępne do analizy białka. Analizie tej poddano także świeżo mrożone tkanki od czterech pozostałych pacjentów autystycznych oraz sześciu pacjentów z grupy kontrolnej, gdzie dostępna była jedynie mrożona tkanka. Tkanki z MFG i ACG były dostępne w 9 z 11 utrwalonych mózgów pacjentów z autyzmem. Świeże tkanki zostały zatopione w parafinie, a następnie skrawki o grubości 10 µm zostały pobrane do badań histologicznych oraz immunocytochemicznych. Próbki mrożonych tkanek z CBL, MFG i ACG w mózgach pacjentów autystycznych (n = 7) i w grupie kontrolnej (n = 7) homogenizowano w buforze lizującym z trzema detergentami zawierającym 50nM Tris-HCl (pH 7.4), 150nM NaCl, 0.02% azydku sodu, 0.1% laurylosiarczanu sodu, 1% odczynnika Igepal (dostawca: Sigma-Aldrich, Inc., St. Louis, MD), 0.5% deoksycholanu sodu i koktajl inhibitorów proteaz (0.2U / ml aprotyniny, 100 g/ml fluorku fenylometylosulfonylu), po czym odwirowano w temperaturze 4°C i złożono do przechowania w temperaturze -80°C. Całkowite stężenie białek obliczono przy użyciu zestawu do badania białek BCA (dostawca: Pierce, Rockford, Illinois), stosując się do szczegółowej procedury opisanej w tym zestawie.
Płyn mózgowo-rdzeniowy
Próbki płynu mózgowo-rdzeniowego [CSF] sześciu żywych pacjentów z autyzmem (w wieku 3-10 lat) zostały pobrane metodą nakłucia lędźwiowego przy zastosowaniu analgosedacji, a następnie niezwłocznie zamrożone w temperaturze -80°C i przechowywano je w stanie zamrożonym do czasu wykorzystania do badania białek. Podobnie próbki CSF pacjentów w grupie kontrolnej (w wieku 12-45 lat) uzyskano z repozytorium CSF Wydziału Neurologii Uniwersytetu Johnsa Hopkinsa. W grupie kontrolnej uwzględniono wyłącznie CFS pacjentów bez dowodów na istnienie chorób zapalnych w ośrodkowym układzie nerwowym i procesów patologicznych (patrz Tabela 2).
Wybarwianie immunocytochemiczne
Wybarwianie immunohistochemiczne wykonano metodą wykorzystującą kompleks awidyna-biotyna-peroksydaza zgodnie z ustaloną procedurą lub zaleceniami producentów. Opis przeciwciał pierwszorzędowych i roztworów znajduje się w Tabeli 3.
Badanie ilościowe immunoreaktywności
Oceny astrogleju (kwaśnego białka włókienkowego, ang. glial fibrillary acidic protein [GFAP]) oraz barwienia immunologicznego aktywowanego mikrogleju (ludzki antygen leukocytarny-DR [HLA-DR]) dokonano przy zastosowaniu nieobciążonej[obiektywnej] techniki analizy ilościowej obszaru ułamkowego zajmowanego przez określony typ komórki zgodnie z powyższym opisem.[25,26] Kora mózgowa MFG i ACG, jak również warstwa ziarnista móżdżku (ang. granular cell layer, GCL) oraz istota biała móżdżku zostały obrysowane do badania ilościowego za pomocą mikroskopu video wykorzystującego Stereo Investigator Software (dostawca: MicroBrightfield, Williston, Vermont). Zbiór 30 punktów został systematycznie rozmieszczony w przypadkowych pozycjach, w odstępach 20 μm w granicach każdego obszaru. Sumę punktów padających na interesujące badaczy struktury (np. astroglej i mikroglej) podzielono przez całkowitą liczbę punktów siatki objętych próbą, aby oszacować fragment obszaru zajęty przez określony typ komórki. Ten obszar ułamkowy został zdefiniowany zgodnie z zasadą Delesse’a[25] jako równy obszarowi zajętemu przez ten rodzaj komórki, który się oblicza. Dzięki powyższej metodzie można zmierzyć procent interesującego naukowców obszaru immunoreaktywnego na konkretne przeciwciało. Jedna osoba, będąc zaślepiona na grupy diagnostyczne, wykonała procedurę liczenia.
Tabela 3. Informacje dotyczące przeciwciał
| Przeciwciało | Rodzaj | Epitop/Specyficzność | Roztwór | Źródło |
| GFAP | Poliklonalne | Astrocyty | 1 : 100 | Dako |
| HLA-DR | Monoklonalne | MHC klasy II, aktywowane komórki mikrogleju | 1 : 100 | Dako |
| CD68 | Monoklonalne | Makrofagi, monocyty | 1 : 100 | Dako |
| MRP-8, kalgranulina A | Monoklonalne | Późne/Przewlekłe nacieki makrofagów | 1 : 100 | BACHEM |
| CD3 | Poliklonalne | Limfocyty T | 1:50 | Dako |
| CD20 | Monoklonalne | Limfocyty B | 1:200 | Dako |
| C9neo (klonu B7) | Monoklonalne | Dopełniacz, kompleks atakujący błonę | 1:20 | Dr P. Morgan, UK |
| IL-6 | Poliklonalne | IL-6 | 1:750 | Novus |
| MCP-1 | Poliklonalne | MCP-1 | 1:200 | Peprotech |
| TGF-P1 | Poliklonalne | TGF-P1 | 1:200 | Santa Cruz |
| IGFBP1 | Poliklonalne | IGFBP-1 | 1:200 | Santa Cruz |
GFAP = kwaśne białko włókienkowe; MRP = białko związane z czynnikiem hamującym migrację makrofagów (MIF) (ang. migration inhibitory factor(MIF)-related protein); IL = interleukina; MCP = białko chemotaktyczne dla makrofagów; TGF = nowotworowy czynnik wzrostu; IGFBP = białko wiążące insulinopodobny czynnik wzrostu.
Mikroskopia konfokalna
Utrwalone w formalinie tkanki mózgu ochroniono krioprotektantem z zastosowaniem roztworów sacharozy, a następnie pocięto za pomocą mikrotomu na skrawki o grubości 40 μm. Skrawki te inkubowano z przeciwciałami pierwszorzędowymi (GFAP+HLADR) i z odpowiednim przeciwciałem drugorzędowym oznaczonym fluorogenem (Cy3 lub Alexa). Próbki zbadano pod laserowym mikroskopem konfokalnym Zeiss LSM 5.0 (dostawca: Zeiss, Thornwood, Nowy Jork).
Mikromacierze do oznaczania tkanki białkowej – Protein Tissue Arrays
Aby dokładniej scharakteryzować naturę reakcji zapalnych w mózgu pacjentów autystycznych, zbadaliśmy względną ekspresję 79 białek: cytokin związanych z odpornością wrodzoną i nabytą, chemokin oraz czynników wzrostu i różnicowania z wykorzystaniem mikromacierzy białkowej do oznaczania ludzkich cytokin.[27,28] Zastosowane zostały zestawy do oznaczania ludzkich cytokin (5.1 i V; dostawca: Raybiotech, Norcros, Georgia), składające się z 79 różnych cytokin, chemokin i czynników wzrostu (Tabela 4) odbitych na błonie nitrocelulozowej. Sposób przeprowadzenia badania odpowiadał instrukcjom producenta: błony zablokowano na godzinę i inkubowano z 500μg homogenatu ludzkiej tkanki bądź 1m CSF przez 2 godziny w temperaturze pokojowej, a następnie płukano przez 30 minut i inkubowano przez 2 godziny w roztworze koktajlu z przeciwciał skoniugowanych z biotyną o stężeniu 1:250. Po kolejnych płukankach dodany został roztwór peroksydazy skoniugowanej ze streptawidyną o stężeniu 1:1000, który inkubowano przez godzinę w temperaturze pokojowej. Błony dokładnie wypłukano i poddano działaniu substratu peroksydazy (chemiluminescencja ECL; dostawca: Amersham, Arlington Heights, Illinois), a następnie zastosowano metodę autoradiografii z przyłożeniem kliszy do błon (Hyperfilm ECL; Amersham) przez czas ekspozycji wynoszący 1 minutę. Klisza została przeskanowana, a punkty digitalizowane na gęstość pikseli z wykorzystaniem oprogramowania do obrazowania NIH (Image J). Współczynnik względnej ekspresji obliczono przez odjęcie intensywności tła i porównanie z dostępnymi w tej błonie białkami rozdzielonymi z początkowej próbki po reakcji z przeciwciałami.
Test ELISA
Nowotworowy czynnik wzrostu (TGF)-p1, białko chemotaktyczne dla makrofagów (MCP)-1, interleukina (IL)-6 (R&D Systems, Minneapolis, Minnesota) i białka wiążące insulinopodobny czynnik wzrostu (IGFBP)-1 (Alpha Diagnostics, San Antonio, Texas) zostały określone w homogenatach tkankowych za pomocą kanapkowego testu ELISA z użyciem dostępnych na rynku zestawów i zgodnie z procedurami ustalonymi przez producentów. Wartości obliczono na podstawie krzywej wzorcowej wygenerowanej dla każdego testu immunoenzymatycznego (ELISA). Próbki rozcieńczono 1 do 10, wyniki ujednolicono zgodnie z ustalonymi wcześniej stężeniami białek, zaś ostateczne stężenia wyrażono w pikogramach na mikrogram białka.
Tabela 4. Białka uwzględnione w analizie z wykorzystaniem mikromacierzy do oznaczania cytokin.
| Cytokiny | Chemokiny | Czynniki wzrostu i różnicowania |
| IL-2 | ENA-78 | GCSF |
| IL-4 | GRO | GM-CSF |
| IL-5 | GRO- α | IL-3 |
| IL-13 | I-309 | IL-7 |
| IFN- γ | IL-8 | MCSF |
| TGF- β1 | MCP-1 | SCF |
| IL-16 | MCP-2 | EGF |
| TGF- β2 | MCP-3 | IGF-I |
| TGF- β3 | MDC | Ang |
| IL-1 α | MIG | OSM |
| IL-1 β | MIP-1 β | Tpo |
| IL-6 | MIP-18 | VEGF |
| IL-10 | RANTES | PDGF-B |
| IL-12 | SDF-1 | Leptyna |
| IL-15 | TARC | BDNF |
| TNF- α | BLC | FGF-4 |
| TNF- β | Ck β 8-1 | FGF-6 |
| Eotaxin | FGF-7 | |
| Eotaxin-2 | FGF-9 | |
| Eotaxin-3 | Ligand FLT3 | |
| Fractalkine | GDNF | |
| GCP-2 | HGF | |
| IP-10 | IGFBP-1 | |
| MCP-4 | IGFBP-2 | |
| MIF | IGFBP-3 | |
| MIP-3 α | IGFBP-4 | |
| NAP-2 | LIF | |
| LIGHT | ||
| NT-3 | ||
| NT-4 | ||
| Osteoprotegeryna | ||
| PARC | ||
| PIGF | ||
| TIMP-1 | ||
| TIMP-2 |
Analiza statystyczna
Do przeprowadzenia wszystkich analiz statystycznych użyto oprogramowania SPSS w wersji 11.0. Ze względu na nieparametryczny charakter danych (co wykazały testy normalności) posłużono się testami nieparametrycznymi dla zwiększenia wiarygodności wyników. Różnice w grupach pomiędzy pacjentami autystycznymi a pacjentami w grupie kontrolnej na obszarze ułamkowym immunoreaktywności na astroglej i aktywowane komórki mikroglejowe w różnych obszarach mózgu zostały porównane z użyciem testu Manna-Whitneya-Wilcoxona z uwagi na, jak się wydawało, niegaussowski charakter danych. Test Manna-Whitneya-Wilcoxona zastosowano także do porównania różnic między grupami w wynikach analizy z użyciem mikromacierzy do oznaczania tkanki białkowej i oznaczania ilościowego testu ELISA. Istotność oceniono na poziomie 0.05. W celu umożliwienia dokonania porównań wielu testów zastosowano poprawkę Bonferroniego, a korelację oceniono za pomocą współczynnika korelacji rang Spearmana z uwagi na porządkowy charakter danych. Testów tych użyto, ponieważ nie robi się w nich założenia na temat rozmieszczenia danych (np. normalności).
Wyniki
W pobranych pośmiertnie mózgach pacjentów z autyzmem obserwuje się wzmożoną aktywację mikrogleju i astrogleju
Nasza analiza zmian neuropatologicznych w tkankach mózgu pacjentów autystycznych wykazała rozległe odpowiedzi komórek glejowych charakteryzujące się aktywacją mikrogleju i astrogleju. W mózgach pacjentów z autyzmem najbardziej wyraźne zmiany histologiczne zaobserwowano w móżdżku, a charakteryzowały się one niejednolitą utratą neuronów w warstwie komórek Purkiniego (ang. Purkinje cell layer, PCL) oraz GCL w 9 spośród 10 móżdżków (Ryc. 1); jeden z tych mózgów wykazał również niemal całkowitą utratę komórek Purkiniego w warstwie komórek Purkiniego [PCL], a także wyraźną utratę komórek ziarnistych (Pacjent 3711, 25-letni mężczyzna chory na epilepsję, patrz Ryc. 1B-D). Tylko w jednym móżdżku nie znaleziono dowodów na utratę komórek Purkiniego (Pacjent 2004, 8-letni chłopiec, patrz Tabela 1). Tymczasem w żadnym obszarze mózgu pacjentów w grupie kontrolnej nie zaobserwowano żadnych znaczących zmian histologicznych. W porówaniu z normalną grupą kontrolną, barwienie immunologiczne GFAP we wszystkich tych obszarach mózgu pacjentów autystycznych wykazało wzmożone odpowiedzi komórek astrogleju charakteryzujące się wzrostem perikarionów i wypustek komórek glejowych. W mózgach pacjentów z autyzmem barwienie immunologiczne GFAP móżdżków wykazało znaczną reaktywność astrogleju w obszarach, gdzie w PCL nastąpiła utrata komórek Purkiniego, jak również znaczącą reakcję astrogleju w GCL oraz istocie białej móżdżku (patrz Ryc. 1G-I). W MFG i ACG reakcje astrogleju były widoczne w podkorowej istocie białej, a w niektórych przypadkach zaobserwowano astrocytozę korową (Ryc. 2). Ocena ilościowa immunoreaktywności astrogleju metodą obszaru ułamkowego[25,26] wykazała znaczny wzrost immunoreaktywności GFAP w takich komorach móżdżku, jak GCL (p < 0.001) i istota biała (p = 0.007) (patrz Ryc. 2I), ale nie osiągnął on statystycznej istotności w MFG (p = 0.076) ani ACG (p = 0.119). Aktywacja astrogleju i reaktywność zostały dokładniej zbadane metodą Western blottingu ekspresji GFAP w homogenatach białkowych pobranych z podgrupy pacjentów autystycznych (n = 7) i kontrolnych (n = 7), od których pozyskano świeżo zamrożoną tkankę mózgu (patrz Tabela 1). Plamy te wykazały znacznie zwiększoną ekspresję GFAP w móżdżku (p = 0.007) i ACG (p = 0.038) pacjentów z autyzmem w porównaniu z pacjentami w grupie kontrolnej.
Aktywację komórek mikrogleju w mózgach pacjentów autystycznych scharakteryzowano ponadto metodą barwienia immunocytochemicznego na markery głównego układu zgodności tkankowej (MHC) klasy II (HLA-DR). Znaczną aktywację mikrogleju zaobserwowano w móżdżku (patrz Ryc. 1C, E-G), w okolicach kory mózgowej (patrz Ryc. 2A, E, F) oraz w istocie białej pacjentów autystycznych. Najbardziej wyraźna reakcja komórek mikrogleju została zaobserwowana w móżdżku, gdzie immunoreaktywność na HLA-DR wykazała znacznie większy obszar ułamkowy immunoreaktywności zarówno w GCL (p < 0.001), jak i w istocie białej móżdżku (p < 0.001) pacjentów z autyzmem niż u pacjentów w grupie kontrolnej (patrz Ryc. 2J). Różnice w aktywacji komórek mikrogleju w MFG (p = 0.106) i ACG (p = 0.109) nie osiągnęły statystycznej istotności. W móżdżku zauważono sporadyczne grudki mikrogleju w GCL oraz istocie białej. Dalsze badania immunocytochemiczne, w tym mikroskopia konfokalna, wykazały, że reakcje zarówno mikrogleju, jak i astrogleju w móżdżku były ściśle związane ze zwyrodniałymi komórkami Purkiniego, komórkami ziarnistymi oraz aksonami (patrz Ry.c 1f, g). W MFG i ACG aktywacja mikrogleju była wyraźna na połączeniu kory mózgowej i istoty białej, a w czterech z dziewięciu przypadków zauważono także umiejscowienie w korze mózgowej. Poza aktywowanymi komórkami mikrogleju zaobserwowaliśmy znaczne skupisko makrofagów okołonaczyniowych oraz monocytów w móżdżkach 4 z 10 pacjetów autystycznych, gdy użyliśmy przeciwciał rozpoznających CD68 (patrz Ryc. 1K) lub antygeny białka 8 (MRP-8) związane z czynnikiem hamującym migrację makrofagów (MIF, ang. migration inhibitory factor), markerów monocytów i makrofagów na etapach przewlekłego stanu zapalnego. Nie zaobserwowaliśmy różnic w aktywacji mikrogleju i astrogleju w zależności od wieku ani profilu klinicznego łącznie z regresją w wywiadzie i upośledzeniem umysłowym pacjentów z autyzmem. Obecność aktywacji mikrogleju w istocie białej móżdżku pacjentów autystycznych z epilepsją w wywiadzie zdaje się znacząco zwiększona (p = 0.025) w porównaniu do pacjentów bez epilepsji, jednak nie zaobserwowano żadnych zmian w GCL ani innych obszarach. Skala reakcji komórek astrogleju zmierzona za pomocą ułamka immunoreaktywnego obszaru bądź metodą western blottingu była podobna w tkankach mózgu pacjentów autystycznych u przypadków z i bez epilepsji w wywiadzie.
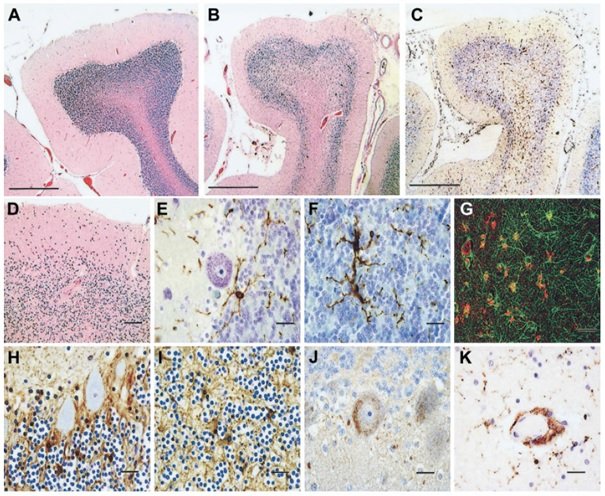
Ryc. 1. Patologia móżdżku w autyzmie. (A) Normalne zakręty móżdżku w mózgu pacjenta w grupie kontrolnej (barwienie na obrazach H i E). (B) W zakrętach móżdżku pacjenta z autyzmem widoczna niejednolita utrata warstwy komórek Pukiniego (PCL) i komórek ziarnistych (GCL), neuronów (H i E) oraz (C) znaczna aktywacja komórek mikrogleju (wybarwionych immunologicznie przeciwciałem anty-HLA-DR). (Kreska w lewym dolnym rogu obrazów A-C = 500 μm. (D) Fragment obszaru móżdżku w znacznym powiększeniu z widoczną utratą neuronów w PCL i GCL (H i E). Kreska w prawym dolnym rogu = 50 μm. (E,F) Aktywowane komórki mikrogleju wokół komórki Purkiniego (E) i w GCL (F) wybarwione immunologicznie przeciwciałem anty-HLA-DR. Kreska w prawym dolnym rogu obrazów E i F = 20 μm. (G) Ścisły związek reaktywnych komórek astrogleju (kolor zielony) i aktywowanych komórek mikrogleju (kolor czerwony) w GCL móżdżku widoczny dzięki zastosowaniu techniki podwójnego barwienia immunocytochemicznego na kwaśne białko włókienkowe (GFAP) (kolor zielony) i HLA-DR (kolor czerwony) i obrazowania za pomocą laserowego mikroskopu konfokalnego (H, I). Zwiększona liczba komórek Bergmanna wokół komórek Purkiniego w PCL na obrazie H i astrocytoza reaktywna w GCL na obrazie I, gdzie zarówno astroglej, jak i astrocytozę wybarwiono immunologicznie przeciwciałami anty-GFAP. (J) Rozpoznanie kompleksu atakującego błonę metodą barwienia immunocytochemicznego przeciwciałem anty-C9neo (struktura ziarnista) w komórkach Purkiniego i innych sąsiednich komórkach, które wydają się komórkani mikrogleju/makrofagami. (K) Skupisko makrofagów okołonaczyniowych i mikrogleju zidentyfikowane przy użyciu przeciwciał anty-CD68. Kreska w prawym dolnym rogu obrazów H-K = 20 μm.
Brak dowodów na adaptacyjną odpowiedź immunologiczną w mózgu pacjentów z autyzmem
W celu dokładniejszego zbadania reakcji immunopatologicznych związanych z odpornością adaptacyjną w mózgu pacjentów autystycznych, wykonaliśmy badania immunocytochemiczne, aby rozpoznać infiltrację limfocytów T i B oraz osadzanie się immunoglobuliny i dopełniacza jako wskaźniki komórkowej i humoralnej odpowiedzi immunologicznej. Zaobserwowaliśmy kilka sporadycznie występujących komórek okołonaczyniowych CD3+ i CD20+ zarówno w mózgach pacjentów z autyzmem, jak i pacjentów w grupie kontrolnej, ale nie zauważyliśmy dowodu na istnienie infiltracji zapalnej w oponach mózgowo-rdzeniowych, komórkach śródmiąższowych ani w tkance okołonaczyniowej w żadnym obszarze poddanych badaniu mózgów osób chorych na autyzm. Barwienie immunologiczne przeciwciałami rozpoznającymi IgC, IgA oraz IgM nie wykazało osadzania się żadnej z tych immunoglobulin w populacjach komórek nerwowych ani glejowych. W móżdżkach pacjentów autystycznych zaobserwowaliśmy osadzanie się kompleksu atakującego błonę (ang. membrane attack complex, MAC) w układzie dopełniacza w perineuronalnych komorach PCL i GCL stosując barwienie immunologiczne przeciwciałem przeciwko antygenowi29 C9neo (patrz Ryc 1L). Schemat immunoreaktywności wskazywał, że niektóre komórki Purkiniego i komórki o morfologii makrofagopodobnej zostały oznaczone przeciwciałem anty-C9neo.
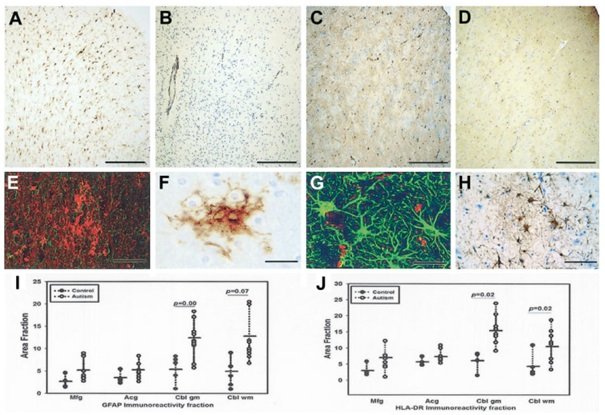
Ryc 2. Reakcje komórek glejowych w korze mózgowej pacjentów z autyzmem. (A-D) Mikroglej aktywowany w korze mózgowej i astrocytoza korowa widoczne w zakręcie czołowym środkowym (MFG) pacjenta autystycznego odpowiednio na obrazach A i C. Na obrazie B widać MFG mózgu pacjenta w grupie kontrolnej wybarwiony immunologicznie na mikroglej, a na obrazie D na astroglej. Barwienie immunologiczne na obrazach A i D przeciwciałami anty-HLA-DR i na obrazach C i D przeciwciałem anty-GFAP. Kreska w prawym dolnym rogu obrazów A-D = 200μm. (E-H) Grudka mikrogleju (E) i skupisko astrocytów reaktywnych (G) w korze mózgowej pacjenta z autyzmem widoczne po podwójnym wybarwieniu immunocytochemicznym na mikroglej (kolor czerwony) i astroglej (kolor zielony) i w obrazowaniu laserem konfokalnym. Podobne skupiska komórek mikroglejowych (F) i astrocytów (H) wizualizowane przy użyciu 3,3-diaminobenzydyny (DAB). (I,J) Obszar ułamkowy immunoreaktywności na GFAP (I) i HLA-DR (J) w zakręcie czołowym środkowym (Mfg), przednim zakręcie obręczy (Acg), PCL i GCL w móżdżku (Cbl gcl) i w istocie białej móżdżku (Cbl-wm). Test Manna-Whitneya-Wilcoxona, poziom istotności p < 0.05.
Podwyższony poziom cytokin prozapalnych w tkankach mózgu pacjentów autystycznych
Oceniliśmy profile ekspresji białek uczestniczących w szlaku zapalnym stosując mikromacierz białkową[27,28] do oznaczania cytokin w homogenatach tkanki mózgu w podgrupie pacjentów autystycznych (n = 7) i kontrolnych (n = 7), od których pobrano świeżo zamrożone tkanki mózgu (patrz Tabela 1). Analiza statystyczna ekspresji względnej cytokin w mózgach pacjentów z autyzmem i pacjentów kontrolnych wykazała ciągły podwyższony poziom podzespołów cytokin w mózgach pacjentów autystycznych (Tabela 1 i Ryc 3): nowotworowy czynnik wzrostu cytokin przeciwzapalnych- β1 (TGF- β1) był wyższy w MFG (p = 0.026), ACG (p = 0.011) i CBL (p = 0.035), a poziom chemokin prozapalnych, MCP-1 i chemokin regulowanych przez aktywację i grasicę (ang. chymus and activation-regulated chemokine, TARC) był podwyższony w ACG (p = 0.026 i 0.035 odpowiednio) i CBL (p = 0.026 i 0.035 odpowiednio). Jedynie poziom IGFBP-1, czynnika wzrostu i różnicowania uczestniczącego w szlaku immunologicznym i szlaku wzrostu komórkowego, był w sposób ciągły podwyższony w obszarach kory mózgowej (MFG, p = 0.038; ACG, p = 0.011), ale różnica ta nie osiągnęła istotności statystycznej w móżdżku (p = 0.11). Co ciekawe, szersze spektrum podwyższonego poziomu cytokin prozapalnych i modulujących zauważono w ACG, gdzie nastąpił znaczący wzrost poziomu interleukin-6 (IL-6), interleukin-10 (IL-10), białka chemotaktycznego dla makrofagów-3 (MCP-2), eotaksyny, eotaksyny 2, chemokin pochodzących z makrofagów (ang. macrophage-derived chemokine, MDC), chemokin-68 (Ckp8.1), peptydów-2 aktywujących neutrofile (ang. neutrophil activating peptide-2, NAP-2), monokin indukowanych przez interferony (MIG), chemoatraktantu limfocytów B (ang. B-lymphocyte chemoattractant, BLC), leptyny i osteoprotegeryny (Ryc 3 i Tabela 5).
Aby potwierdzić nasze obserwacje wynikające z badania przeprowadzonego z użyciem mikromacierzy białkowej do oznaczania cytokin, przeprowadziliśmy test ELISA, za pomocą którego określiliśmy najbardziej istotne cytokiny, TGF- β1, MCP-1, IGFBP-1 oraz IL-g w tej samej grupie tkanek mózgów pacjentów autystycznych i w grupie kontrolnej (Ryc 4 i Tabela 6). Ustaliliśmy, że w trzech badanych obszarach poziom cytokin przeciwzapalnych TGF-β1 był w sposób ciągły i znacząco wyższy w grupie autystycznej niż kontrolnej; poziom/ białek MCP-1 także był znacząco podwyższony w ACG (p = 0.001) pacjentów z autyzmem, a liczby te niemal osiągnęły istotność w MFG (p = 0.057). Podobnie, stężenia IGFBP-1 były znacząco wyższe w MFG (p = 0.032) i ACG (p = 0.01) pacjentów autystycznych, ale nie odkryto żadnej istotnej różnicy w CBL (p = 0.42). Dla porównania, nie odkryliśmy żadnych znaczących różnic stężeń IL-6 w MFG (p = 0.45), ACG (p = 0.12) ani w CBL (p = 0.32) w mózgach pacjentów autystycznych i w grupie kontrolnej.
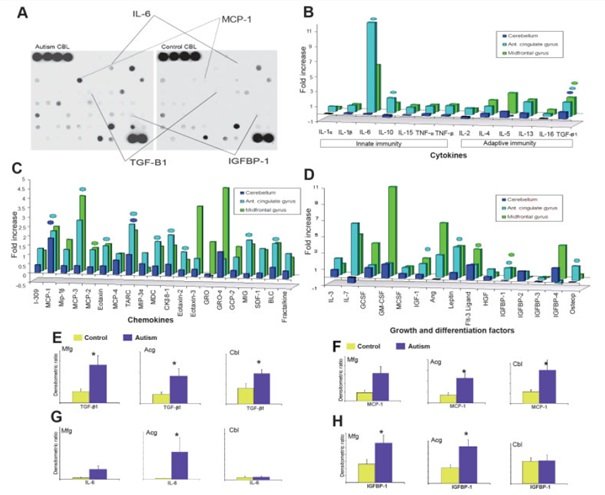
Ryc 3. Profil cytokin w tkance mózgu uzyskany z wykorzystaniem mikromacierzy białkowej do oznaczania cytokin. (A) Dwie membrany macierzy służących do oznaczania cytokin, homogenaty białek z móżdżku pacjenta autystycznego i pacjenta w grupie kontrolnej. Każda kropka oznacza cytokinę, dla której współczynnik ekspresji (jednostki miary kąta) uzyskano pomiędzy tą cytokiną a kontrolą pozytywną obecną w tej błonie. Umiejscowienie w błonie białka chemotaktycznego dla makrofagów (MCP)-1, interleukin (IL)-6, nowotworowego czynnika wzrostu (TGF)-β1 oraz IGFBP-1 przedstawiono w obu błonach dla porównania. (B-D) Profile ekspresji (B) cytokin, (C) chemokin i (C) czynników wzrostu i różnicowania w móżdżku (Cbl), przednim zakręcie obręczy (Acg) i zakręcie czołowym środkowym (Mfg). (E-H) Współczynnik ekspresji (jednostki miary kąta) TGF-β1, MCP-1, IGFBP-1 w różnych obszarach mózgu pacjentów autystycznych i w grupie kontrolnej. Istotność statystyczną (*) uzyskano na poziomie p < 0.05 na podstawie testu Manna-Whitneya-Wilcoxona.
Astrocyty reaktywne głównym źródłem cytokin w mózgach pacjentów z autyzmem
W celu określenia źródeł komórkowych najistotniejszych cytokin w mózgach pacjentów autystycznych, wykonaliśmy barwienie na TGF-β1, MCP-1, IGFBP-1 oraz IL-6 w MFG, ACG i CBL. Zaobserwowane wzory barwienia wskazywały, że astrocyty stanowiły główne źródło zarówno MCP-1, jak i IL-6. Ekspresja obu cytokin była wyraźnie zwiększona w astrocytach reaktywnych w móżdżku oraz w istocie białej kory mózgowej i w podkorowej istocie białej. Analiza pod mikroskopem konfokalnym skrawków podwójnie wybarwionych immunologicznie na GFAP i MCP-1 lub GFAP i IL-6 dodatkowo potwierdziła te obserwacje, a także współwystępowanie tych cytokin w astrocytach (patrz Ryc 2). Warto zwrócić uwagę, że ekspresję TGF-β1 oraz IGFBP-1 zauważono nie tylko w astrocytach reaktywnych, ale również w populacji komórek Purkiniego i w podgrupach komórek GCL w CBL. Niektóre komórki mikrogleju zostały także oznakowane przeciwciałami rozpoznającymi TGF-β1 i IGFBP-1. Komórki Purkiniego ze zmianami zwyrodnieniowymi okazały się silnie immunoreaktywne na TGF-β1 (patrz Ryc 4).
Tabela 5. Podwyższony poziom cytokin w tkankach mózgu pacjentów z autyzmem
| Zakręt czołowy środkowy | Przedni zakręt obręczy | Móżdżek | ||||
| Cytokina | Krotne zwiększenie | pa | Krotne zwiększenie | pa | Krotne zwiększenie | pa |
| IL-6 | 6.10 | nieistotne | 31.4 | 0.011 | 0.11 | nieistotne |
| IL-10 | 1.11 | nieistotne | 2.00 | 0.007 | -0.33 | nieistotne |
| TGF-β1 | 2.33 | 0.026 | 2.00 | 0.011 | 0.92 | 0.035 |
| MCP-1 | 2.32 | nieistotne | 2.20 | 0.026 | 1.90 | 0.035 |
| MCP-2 | 1.24 | 0.017 | 1.20 | nieistotne | 0.37 | nieistotne |
| MCP-3 | 4.00 | nieistotne | 2.80 | 0.038 | 0.42 | nieistotne |
| Eotaksyna | 1.46 | nieistotne | 1.40 | 0.017 | 0.44 | nieistotne |
| TARC | 2.33 | nieistotne | 2.63 | 0.026 | 1.09 | 0.035 |
| MDC | 1.41 | nieistotne | 1.74 | 0.004 | 0.51 | nieistotne |
| CKβ8.1 | 1.47 | nieistotne | 2.11 | 0.017 | 0.62 | nieistotne |
| Eotaksyna 2 | 0.73 | nieistotne | 1.22 | 0.038 | 0.34 | nieistotne |
| MIG | 1.65 | nieistotne | 1.94 | 0.026 | 0.22 | nieistotne |
| BLC | 1.75 | nieistotne | 1.80 | 0.026 | 0.60 | nieistotne |
| IGF-1 | 2.09 | nieistotne | 1.63 | 0.026 | 0.51 | nieistotne |
| Leptyna | 3.72 | nieistotne | 3.8 | 0.007 | 0.84 | nieistotne |
| Ligand Flt3 | 3.34 | nieistotne | 1.71 | nieistotne | 1.86 | 0.022 |
| IGFBP-1 | 1.14 | 0.038 | 1.36 | 0.011 | 0.02 | nieistotne |
| Osteoprotegeryna | 0.057 | nieistotne | 1.78 | 0.017 | 0.03 | nieistotne |
a Test Manna-Whitneya-Wilcoxona.
IL = interleukina; TGF = nowotworowy czynnik wzrostu; MCP = białko chemotaktyczne dla makrofagów; MDC = chemokina pochodząca z makrofagów; insulinopodobny czynnik wzrostu IGFBP = białko wiążące IGF.
Płyn mózgowo-rdzeniowy pacjentów autystycznych wykazuje profil prozapalny
Jako że tkanki mózgu pacjentów z autyzmem wykazywały znaczny profil prozapalny, wzięliśmy pod uwagę możliwość, że CSF tych pacjentów może mieć podobny profil. Mikromacierze białkowe do oznaczania cytokin wykorzystano do porównania profili cytokinowych CSF sześciu pacjentów autystycznych z profilami cytokinowymi CSF pochodzącymi od banku dawców bez patologii CNS i zaburzeń zapalnych (np. rzekomego guza mózgu czy bólów głowy; patrz Tabela 2). Tak jak zaobserwowaliśmy w tkance mózgu, płyn mózgowo-rdzeniowy [CSF] pacjentów z autyzmem wykazał istotny wzrost poziomu MCP-1 (12-krotny) w porównaniu z pacjentami w grupie kontrolnej (Tabela 7 i Ryc 5). Nie wystąpiły różnice w ekspresji TARC ani TGF-β1 w CSF. Jednakże poziom innych cytokiny prozapalnych i modulacyjnych, takich jak IL-6, interferon (IFN-γ), IL-8, białko zapalne makrofagów-1β (ang. macrophage inflammatory protein-1β, MIP1β), białko aktywujące neutrofile (ang. NAP-2), białko indukowane interferonem-γ (ang. interferon-γ induced protein-10, IP-10) i angiogenina, a także czynniki wzrostu, w tym czynnik indukujący mezodermę (ang. mesoderm inducing factor, MIF), czynnik wzrostu śródbłonka naczyniowego (ang. vascular endothelial growth factor, VEGF), czynnik hamujący białaczkę (ang. leukemia inhibitory factor, LIF), osteoprotegeryna, wątrobowy czynnik wzrostu (ang. hepatic growth factor, HGF), PARC, FGF-4, FGF-9, IGFBP3 oraz IG-FBP4 był znacząco podwyższony w porównaniu z CSF w grupie kontrolnej (patrz Ryc 5 i Tabela 7).
Omówienie/dyskusja
Reakcje mikrogleju i astrogleju u chorych na autyzm charakteryzują wrodzone odpowiedzi immunologiczne
W badaniu tym przedstawiliśmy wyraźne nasilenie odpowiedzi komórek glejowych w mózgach pacjentów autystycznych, charakteryzujące się aktywacją mikrogleju i astrogleju. Takie nasilone odpowiedzi komórek glejowych stanowi prawdopodobnie część reakcji zapalnej układu nerwowego związanej z wrodzonym układem odpornościowym Ośrodkowego Układu Nerwowego [CNS], w którym aktywacja mikrogleju jest główną odpowiedzią komórkową na dysfunkcję Ośrodkowego Układu Nerwowego[30,31] w porównaniu z nabytymi odpowiedziami immunologicznymi, w których reakcje z udziałem limfocytów lub przeciwciał są odpowiedziami dominującymi.[32,33] W naszej próbie statystycznej przypadków autystycznych, aktywacja komórek mikrogleju i astrogleju była obecna tam, gdzie nie występowała infiltracja limfocytów ani odkładanie się immunoglobulin w Ośrodkowym Układzie Nerwowym [CNS], i łączono ją ze zwiększoną produkcją cytokin prozapalnych i przeciwzapalnych takich jak MCP-1 i TGF-1 przez komórki glejowe.
Ponieważ autyzm jest zaburzeniem heterogenicznym, które można łączyć z licznymi przyczynami, możliwe, że nasza próba statystyczna przypadków nie reprezentuje całego spektrum autystycznego, jako że niektórzy z naszych pacjentów mieli inne powiązane zaburzenia neurologiczne często towarzyszące autyzmowi, takie jak epilepsja i upośledzenie umysłowe.
Jednak obecność odkryć morfologicznych oraz immunologicznych wskazujących na reakcje neuroimmunologiczne u próby statystycznej pacjentów z autyzmem objętych tym badaniem, jak również wykrycia w płynie mózgowo-rdzeniowym wspierają potencjalną rolę komórek glejowych i stanu zapalnego w układzie nerwowym jako mechanizmów patogenicznych u nieokreślonego odsetka osób chorych na autyzm.
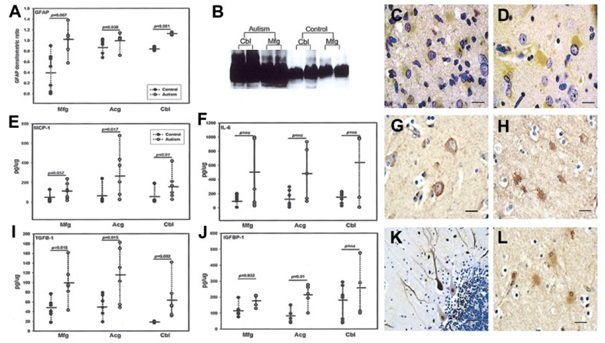
Ryc 4. Weryfikacja za pomocą analizy ilościowej przeprowadzona przy użyciu testu ELISA oraz immunolokalizacja ekspresji cytokin w mózgu. (A) Ekspresja kwaśnego białka włókienkowego (GFAP) określona metodą immunoblotingu (jednostki arbitralne) była znacząco zwiększona we wszystkich obszarach mózgu pacjentów z autyzmem. (B) Obraz z badania GFAP w móżdżku (Cbl) i w zakręcie czołowym środkowym (MFG) pacjentów autystycznych i w grupie kontrolnej metodą western blottingu. (C) Białko chemotaktyczne dla makrofagów (MCP) – 1 immunolokalizacja w astrocytach.
(D) Interleukina (IL)-6 została wykryta immunochemicznie w astrocytach reaktywnych. (E) Stężenie MCP-1 było znacząco większe w Acg i Cbl przypadków z autyzmem. (F) Poziom IL-6 był podwyższony we wszystkich trzech obszarach, ale jej wzrost nie osiągnął istotności statystycznej. (G) IGFBP-1 została zlokalizowana w podgrupach neuronów kory mózgowej, komórkach Purkiniego i (H) astrocytach reaktywnych. (I) Kwantyfikacja TGF-β1 i (j) IGFBP-1. (K) TGF-β1 zlokalizowano również w komórkach Purkiniego (L), astrocytach reaktywnych i neuronach kory mózgowej (nie pokazano). Podzespoły komórek Purkiniego ze zmianami morfologicznymi zgodnymi ze zwyrodnieniem zostały intensywnie wybarwione immunologicznie przeciwciałami anty-TGF-β1. Istotność statystyczną uzyskano/ na poziomie p < 0.05 na podstawie testu Manna-Whitneya-Wilcoxona.
Tabela 6. Analiza ilościowa ELISA wybranych cytokin w homogenatach tkanki mózgowej
| MCP1 (pg/μg) | IL-6 (pg/μg) | TGF-β1 (pg/μg) | IGFBP-1 (pg/μg) | |||||
| Obszar mózgu | Średnia | SEM | Średnia | SEM | Średnia | SEM | Średnia | SEM |
| Móżdżek | ||||||||
| Grupa autystyczna | 163.6 | 57.94 | 618.4 | 221.9 | 64.17 | 16.75 | 244.8 | 88.74 |
| Grupa kontrolna | 38.51 | 23.74 | 137.5 | 119.8 | 18.37 | 0.57 | 161.1 | 60.71 |
| MFG | ||||||||
| Grupa autystyczna | 110.8 | 30.05 | 482.7 | 180.3 | 98.62 | 15.96 | 210.6 | 27.95 |
| Grupa kontrolna | 36.77 | 14.8 | 130.7 | 26.48 | 47.11 | 8.24 | 85.89 | 18.65 |
| ACG | ||||||||
| Grupa autystyczna | 260.6 | 93.15 | 492.6 | 171.6 | 114.8 | 23.18 | 181.43 | 17.06 |
| Grupa kontrolna | 50.82 | 29.67 | 138.9 | 48.99 | 53.35 | 9.42 | 118.9 | 21.23 |
ELISA = test immunoenzymatyczny; MCP = białko chemotaktyczne dla makrofagów; IL = interleukina; TGF = nowotworowy czynnik wzrostu; IGFBP = białko wiążące insulinopodobny czynnik wzrostu; SEM = błąd standardowy średniej, ang. standard error of the mean); MFG = zakręt czołowy środkowy; ACG = przedni zakręt obręczy
Tabela 7. Cytokiny, których poziom wzrósł znacząco w płynie mózgowo-rdzeniowym pacjentów z autyzmem
| Cytokina | Krotne zwiększenie | pa |
| IFN-7 | 232.5 | 0.008 |
| TGF-β2 | 30.9 | <0.001 |
| MCP-1b | 12.2 | <0.001 |
| IL-8 | 6.0 | <0.001 |
| IP-10 | 18.2 | 0.018 |
| Angiogeninab | 3.3 | 0.003 |
| VEGF | 81.8 | 0.001 |
| IGFBP-1 | 0.4 | 0.036 |
| IGFBP-3 | 26.3 | <0.001 |
| IGFBP-4 | 13.3 | 0.003 |
| LIF | 1.0 | <0.001 |
| FGF-4 | 0.23 | 0.005 |
| FGF-9 | 70.0 | 0.012 |
| PARC | 11.3 | 0.002 |
| Osteoprotegerynab | 5.2 | 0.002 |
| HGF | 0.3 | 0.005 |
| IGFBP-3 | 26.3 | <0.001 |
| IGFBP-4 | 13.3 | 0.003 |
a Test Manna-Whitneya-Wilcoxona.
b Odkryto znaczący wzrost także w jednym lub większej liczbie obszarów w badaniu tkanki mózgu.
IFN = interferon; TGF = nowotworowy czynnik wzrostu; MCP = białko chemotaktyczne dla makrofagów; IL = interleukina; VEGF = czynnik wzrostu śródbłonka naczyniowego; IGFBP = białko wiążące insulinopodobny czynnik wzrostu; LIF = czynnik hamujący białaczkę; HGF = wątrobowy czynnik wzrostu.
Aktywacja komórek glejowych w tkance mózgu pacjentów autystycznych była szczególnie uderzająca w móżdżku, a zmiany te były związane z regulacją w górę wybiórczych cytokin w tym i innych obszarach mózgu. Analiza immunocytochemiczna reakcji mikrogleju i astrogleju w mózgach tych pacjentów wykazała, że niezależnie od wieku, epilepsji w wywiadzie, regresu w rozwoju czy upośledzenia umysłowego, wyraźne zmiany morfologiczne wskazujące na przewlekłe i ciągłe odpowiedzi zapalne komórek glejowych były obecne w istocie białej kory mózgowej i w podkorowej istocie białej, a także w móżdżku. Zmiany te mogą uczestniczyć w mechanizmach związanych z dysfunkcją neuronową i synapatyczną występującą w autyzmie. Komórki mikrogleju, makrofagi rezydentne i komórki immunokompetentne układu nerwowego[30,34] były konsekwentnie aktywowane we wszystkich obszarach mózgu pacjentów autystycznych, ale zwłaszcza w móżdżku. Podobnie, badanie metodą western blottingu wskazało na zwiększoną ekspresję GFAP (wskaźnik skali aktywacji komórek astrogleju) we wszystkich zbadanych obszarach w porównaniu z normalnymi przypadkami w grupie kontrolnej.
Reakcje komórek mikrogleju u pacjentów z autyzmem były rozmieszczone w rozproszeniu w obszarze kory mózgowej i obszarze podkorowym, jak również w móżdżku, lub też były obecne w postaci grudek komórek mikrogleju bądź jako część wyraźnego skupiska makrofagów okołonaczyniowych. Odpowiedzi te w autyzmie przypominają odpowiedzi obserwowane w chorobach neurodegeneracyjnych takich jak choroba Alzheimera[26] (ang. Alzherimer’s disease, AD), choroba Parkinsona (ang. Parkinson’s disease, PD) i stwardnienie zanikowe boczne,[35-37] a także zbliżone są do tych obserwowanych w demencji związanej z infekcją ludzkim wirusem niedoboru odporności (ang. human immunodeficiency virus, HIV).[38,39] W chorobach tych, przewlekła aktywacja komórek mikrogleju wydaje się odpowiadać za ciągłą odpowiedź zapalną komórek nerwowych umożliwiającą produkcję licznych mediatorów neurotoksycznych.[40,41] Aktywacja stanu zapalnego układu nerwowego może być uniwersalną ścieżką prowadzącą do dysfunkcji CNS we wszystkich tych chorobach. W przypadku autyzmu aktywacja komórek mikrogleju potwierdza pogląd, że wrodzone odpowiedzi immunologiczne są obecne w obszarze kory mózgowej i w obszarze podkorowym oraz że stan przewlekłej aktywacji i reaktywności może wchodzić w skład mechanizmów dysfunkcji neuronowej i synaptycznej.
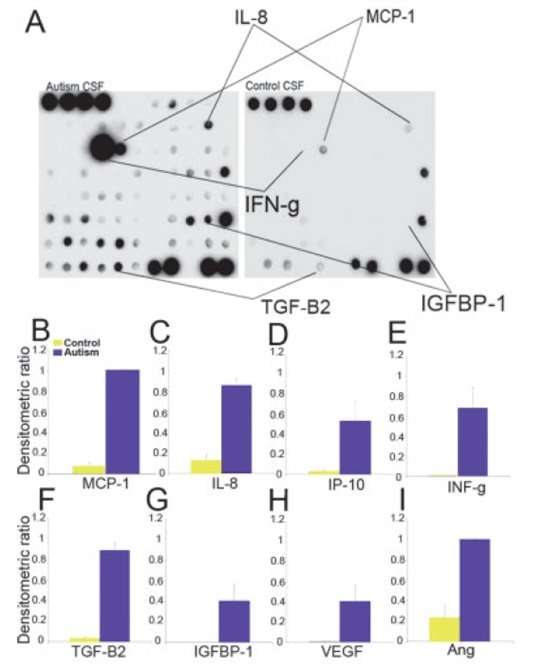
Ryc. 5 Profile cytokin w płynie mózgowo-rdzeniowym (CSF) pobranym od pacjentów austystycznych i kontrolnych. (A) Mikromacierze białkowe do oznaczania cytokin próbki CSF pobrane od pacjentów z autyzmem i pacjentów w grupie kontrolnej. Punkty oznaczające białko chemotaktyczne dla makrofagów (MCP)-1, interferon-γ, TGF-β2, interleukinę-8 oraz IGFBP-1 wykazały wyraźny wzrost gęstości w porównaniu z CSF pobranym od pacjentów w grupie kontrolnej. (B-I) Profil ekspresji (krotny wzrost) cytokin, których poziom wyraźnie się zwiększył u pacjentów autystycznych w porównaniu z pacjentami w grupie kontrolnej. p < 0.05, test Manna-Whitneya-Wilcoxona.
Obecność zwiększonych odpowiedzi komórek glejowych ma znaczenie dla mechanizmów neurobiologicznych występujących w autyzmie, ponieważ zarówno komórki mikrogleju, jak i astrogleju są niezbędne dla funkcjonowania neuronów i aktywności synaptycznej,[42] interakcji między neuronami a komórkami glejowymi, jak również dla budowy, organizacji i przebudowy kory mózgowej w procesie rozwoju mózgu.[44] Ponadto aktywacja komórek mikrogleju i astrogleju wydaje się odgrywać główną rolę w mechanizmach neuroimmunologicznych chorób CNS,[34] ponieważ komórki te są częścią pierwszej linii obrony wrodzonego układu odpornościowego CNS[30] i przyczyniają się do modulowania odpowiedzi immunologicznej wytwarzając zarówno cytokiny prozapalne, jak i przeciwzapalne, a także czynniki wzrostu i różnicowania.[45] Odpowiedzi komórek mikrogleju i astrogleju w CNS mogą zatem pełnić dwojaką rolę w odpowiedziach zapalnych mózgu: jako bezpośredni efektor uszkodzenia i z drugiej strony jako neuroprotektant.[46] Kwestią, która pozostaje niejasna jest to jak i kiedy dochodzi do aktywacji komórek mikrogleju i astrogleju w mózgu pacjentów z autyzmem. Odpowiedzi komórek glejowych w autyzmie mogą być częścią zarówno pierwotnych (wewnętrznych) odpowiedzi tych komórek będących skutkiem zaburzeń ich funkcjonowania lub interakcji między tymi komórkami a neuronami w procesie rozwoju mózgu, jak i wtórnych (zewnętrznych) powodowanych przez nieznane czynniki, które zakłócają prenatalny albo ponatalny rozwój CNS. Zarówno astrocyty, jak i komórki mikrogleju są kluczowe dla rozwoju mózgu i MHC klasy II (antygen HLA-DR) – dodatnie komórki mikrogleju zasiedlają rozwijający się CNS w drugim trymestrze ciąży.[47,48] Możliwe, że obecność aktywowanych komórek mikrogleju w mózgu pacjentów autystycznych odzwierciedla anormalną ciągłość wzorców rozwoju płodu w odpowiedzi na czynniki genetyczne lub środowiskowe (np. wewnątrzmaciczne czy związane z matką). Pomimo że nasze analizy nie wykazały żadnej różnicy między aktywacją komórek glejowych wśród pacjentów autystycznych z regresem w rozwoju bądź upośledzeniem umysłowym w wywiadzie, do wyjaśnienia tych kwestii potrzebne są dalsze badania obejmujące większe grupy przypadków.
Wcześniejsze badania neuropatologiczne autyzmu wykazały nieprawidłowości w budowie kory mózgowej i upakowaniu neuronów, a także mniejszą liczbę komórek Purkiniego w móżdżku.[11] Nasze ustalenia mogą wskazywać, że w którymś momencie podczas powstawania struktury kory mózgowej i komórek nerwowych, nieznane czynniki wpływają zarówno na populację neuronów, jak i komórek glejowych, zaburzając rozwój neurologiczny i powodując zmiany cytoarchitektoniki układu nerwowego obserwowane u chorych na autyzm, jak również indukując dysfunkcję CNS skutkującą stanem zapalnym układu nerwowego. Alternatywą dla tego wyjaśnienia jest możliwość, że zewnętrzne przyczyny (np. niegentyczne, neurotoksyczne lub środowiskowe) związane z patogenezą autyzmu mogą powodować anomalie komórek nerwowych i kory mózgowej, na które reakcje komórek glejowych są jedynie ubocznymi [wtórnymi] odpowiedziami. Pomimo że znaczenie stanu zapalnego układu nerwowego w naszej próbie statystycznej jest w tym momencie nieznane, takie pierwotne lub uboczne[wtórne] odpowiedzi mogą być cennymi klinicznymi biomarkerami oraz celem terapii, o ile możliwe jest dowiedzenie, że powodują uszkodzenia rozwijającego się CNS.
Brak adaptacyjnych odpowiedzi immunologicznych w mózgach pacjentów z autyzmem
W przeciwieństwie do widocznej obecności aktywowanych komórek mikrogleju i astrocytów, właściwości charakteryzujących wrodzoną odpowiedź immunologiczną w CNS, ważnym odkryciem w naszym badaniu był brak specyficznej odpowiedzi limfocytów T oraz reakcji z udziałem przeciwciała w jakimkolwiek zbadanym obszarze mózgu pacjentów autystycznych. Obserwacje te wskazują, że układ odpornościowy nabyty nie odgrywa istotnej patogenicznej roli w tym zaburzeniu, a przynajmniej nie w jego fazie przewlekłej, jak również, że główny mechanizm immunologiczny obejmuje przede wszystkim wrodzoną odpowiedź immunologiczną. Jako że nasze badanie koncentrowało się na tkankach pobranych podczas sekcji zwłok, nie możemy wykluczyć, iż specyficzna odpowiedź immunologiczna z udziałem odpowiedzi limfocytów T i/lub przeciwciał, wystąpiła na początku choroby, na prenatalnym lub postnatalnym etapie rozwoju. Interesujące ustalenie w naszych badaniach immunocytochemicznych dotyczyło obserwacji kompleksu atakującego błonę układu dopełniacza w móżdżkach. Umiejscowienie oraz immunoreaktywność C9neo, markera kompleksu atakującego błonę,[29] w perineuronalnych komórkach Purkiniego i w obszarach centralnych wskazują, że aktywacja komórek mikrogleju może wywoływać aktywację układu dopełniacza i układ ten może odgrywać rolę w destrukcyjnym procesie zachodzącym w móżdżku pacjentów z autyzmem. Jednakże nieodkładanie się immunoglobulin wskazuje, że aktywacja układu dopełniacza może wystąpić w przypadku braku ścieżek zależnych od przeciwciał i może przypominać mechanizmy immunopatogeniczne obserwowane u chorych na AD, PD i inne choroby neurodegeneracyjne, w których zjawiska autotoksyczne odgrywają rolę w uszkodzeniu komórek nerwowych oraz neurodegeneracji.[49] Konieczne jest dalsze wyjaśnienie roli tych reakcji autotoksycznych (z udziałem układu dopełniacza i związanych z inną wrodzoną odpowiedzią immunologiczną w móżdżku), gdyż wydaje się, że degeneracja PCL i GCL zachodzi w przypadku braku adaptacyjnej odpowiedzi immunologicznej.
Móżdżek jest głównym ogniskiem stanu neurozapalnego w autyzmie
Przeprowadzona przez nas analiza ilościowa reakcji komórek glejowych wskazała, że spośród zbadanych obszarów mózgu, móżdżek wykazywał najbardziej wyraźne odpowiedzi komórek glejowych. Ta widoczna aktywność komórek glejowych w móżdżku pokrywa się z wcześniejszymi obserwacjami, że móżdżek jest jednym z siedlisk anomalii patologicznych w badaniach morfologicznych[11,12] i neuroobrazowaniu[52,-52] pacjentów z autyzmem. Na podstawie naszych spostrzeżeń wydaje się, że selektywny proces degeneracji neuronów i aktywacja komórek glejowych występują przeważnie w PCL i GCL móżdżku pacjentów z autyzmem, przy czym ustalenia te wskazują na aktywny i ciągły postnatalny proces neurodegeneracji oraz stan zapalny układu nerwowego. Obserwacje te nie potwierdzają wcześniej wysuwanej hipotezy, że zmiany w móżdżku u chorych na autyzm powstają wyłącznie na skutek nieprawidłowości rozwojowych w układzie oliwkowo-móżdżkowym i redukcji liczby komórek Purkiniego.[11] Z kolei nasze spostrzeżenia wskazują, że zmiany patologiczne obserwowane w móżdżku pacjentów autystycznych nie zachodzą tylko i wyłącznie w okresie rozwoju prenatalnego, ale wydaje się, iż obejmują trwały, przewlekły proces zapalny układu nerwowego, który uwzględnia zarówno komórki mikrogleju, jak i astrogleju. Co więcej, proces ten trwa dłużej niż etap wczesnego rozwoju neurologicznego i występuje nawet w bardzo późnych okresach życia pacjentów z autyzmem. Ustalenia te potwierdzają także hipotezę, że selektywna podatność komórek Purkiniego odgrywa rolę w etiopatogenezie autyzmu.[53]
Białko chemotaktyczne dla makrofagów typu 1 i nowotworowy czynnik wzrostu β1 są najbardziej wyraźnymi cytokinami w mózgu pacjentów autystycznych
Nasze badanie wykazało również obecność unikalnych profili ekspresji cytokin w mózgu i CSF pacjentów z autyzmem. Poziom dwóch chemokin prozapalnych, MCP-1 i TARC, a także cytokiny przeciwzapalnej i modulującej TGF-β1 były konsekwentnie podwyższone w zbadanych obszarach mózgu. Wydaje się, że MCP-1, chemokina uczestnicząca we wrodzonej odpowiedzi immunologicznej i ważny mediator monocytów oraz aktywacji limfocytów T, a także transportu do obszarów uszkodzenia tkanki, jest jednym z najważniejszych białek odkrytym w badaniach z użyciem mikromacierzy białkowej do oznaczania cytokin, ponieważ jej poziom był znacząco podwyższony zarówno w tkankach mózgu, jak i CSF. Szczególne zainteresowanie budzi obecność MCP-1, ponieważ cytokina ta ułatwia naciekanie i nagromadzanie monocytów oraz makrofagów w chorobie zapalnej CNS.[55] Jak wykazały nasze badania immunocytochemiczne kory mózgowej i móżdżku, MCP-1 produkują monocyty aktywowane i reaktywne, zaś ustalenie to uwidacznia rolę efektora, którą komórki te pełnią w procesie chorobowym autyzmu. Zwiększona ekspresja MCP-1 ma znaczenie dla patogenezy autyzmu, gdyż sądzimy, że jej podwyższenie w mózgu wiąże się z aktywacją komórek mikrogleju i być może z pozyskiwaniem monocytów/makrofagów na obszary neurodegeneracji, takie jak zaobserwowane przez nas w móżdżku. Nasze obserwacje przypominają ustalenia dotyczące innych chorób neurologicznych, w których podwyższenie poziomu MCP-1 związane jest z patogenezą stanu zapalnego układu nerwowego i uszkodzeniem komórek nerwowych, takich jak encefalopatia AIDS [HIV dementia],[56] stwardnienie zanikowe boczne,[37] udar[57] i stwardnienie rozsiane.[55] Pozostaje niejasne czy MCP-1 odgrywa bardziej pleotropiczną rolę w CNS i czy jej obecność związana jest wyłącznie ze stanami zapalnymi. Przypuszcza się, że MCP-1 może być zaangażowana w mechanizm przetrwania neuronów i mechanizm neuroprotekcyjny [neuroochronny] inny niż aktywacja i transport monocytów[58] lub nawet w mechanizm uszkodzenia komórek nerwowych bez udziału limfocytów.[59] Wydaje się, że ekspresja MCP-1 w CNS jest regulowana rozwojowo, zaś wcześniejsze badania wykazały ekspresję tej cytokiny w móżdżku w okresie rozwoju prenatalnego, co może wskazywać na związek z dojrzewaniem komórek Purkiniego.[60] Podobnie jak w przypadku ekspresji MHC klasy II w mikrogleju na etapie modelowania CNS,[47] podwyższenie poziomu MCP-1 w mózgach pacjentów autystycznych może odzwierciedlać trwałe wzorce rozwoju mózgu płodu.
Nasza obserwacja, że poziom TG-β1 zwiększył się w korze mózgowej i móżdżku mózgu pacjentów z autyzmem może mieć istotne znaczenie dla neurobiologii autyzmu. TGF-β1 jest kluczową cytokiną przeciwzapalną uczestniczącą w remodelowaniu uszkodzonej tkanki. Może stłumić specyficzną odpowiedź immunologiczną hamując proliferację i dojrzewanie limfocytów T oraz reguluje w dół ekspresję MHC klasy II.[61] Co ważne, wykazano, iż komórki ulegające śmierci komórki wydzielają TGF-β1, prawdopodobnie po to, by złagodzić miejscowy stan zapalny i zapobiec degeneracji dodatkowych sąsiednich komórek.[62] W naszych badaniach immunocytochemicznych, TGF-β1 została zlokalizowana głównie w astrocytach reaktywnych i neuronach w móżdżku. Komórki Purkiniego wykazujące właściwości morfologiczne degeneracji odznaczały się wyraźną immunoreaktywnością na TGF-β1. Ustalenia te wskazują, że podwyższenie poziomu tej cytokiny w autyzmie może odzwierciedlać próbę modulowania stanu zapalnego układu nerwowego lub remodelowania i naprawy uszkodzonej tkanki. Mimo że poziom TGF-β1, MCP-1, TARC oraz IGFBP-1 był konsekwentnie podwyższony w co najmniej dwóch spośród trzech zbadanych obszarów mózgów osób z autyzmem, bardziej godny uwagi profil regulacji w górę cytokin zaobserwowano w ACG, czyli w obszarze, gdzie poziom kilkunastu cytokin, chemokin i czynników wzrostu był wyraźnie podwyższony w porównaniu z grupą kontrolną. Zarówno poziom cytokin prozapalnych (np. IL-6), jak i przeciwzapalnych (np. IL-10), a także podgrup chemokin był wyraźnie podwyższony w ACG, ważnym obszarze kory mózgowej uczestniczącym w dysfunkcyjnej czynności mózgu u chorych na autyzm.[63] Ustalenia te wspierają konkluzję, że aktywny, trwały proces immunologiczny występował w licznych obszarach mózgu, ale w każdym odznaczał się innym poziomem ekspresji.
Wyraźna ekspresja cytokin prozapalnych w płynie mózgowo-rdzeniowym pacjentów z autyzmem
Badania CSF potwierdziły również znaczący profil cytokin zapalnych u pacjentów chorych na autyzm. Widoczny wzrost ekspresji MCP-1 w CSF wspiera hipotezę, że szlaki prozapalne aktywują się w mózgu pacjentów autystycznych i że wzrost ten może się wiązać z mechanizmami aktywacji makrofagów/komórek mikrogleju obserwowanymi w badaniach tkanki mózgu. Podwyższenie ekspresji MCP-1 w CSF przypomina obserwacje w innych schorzeniach, w których aktywacja komórek mikrogleju/makrofagów pełni istotną rolę, jak encefalopatia AIDS[56] czy stwardnienie rozsiane.[64] Oprócz wyraźnego zwiększenia ekspresji MCP-1, podwyższone poziomy interferonu-γ, IL-8, IL-10 i innych cząsteczek prozapalnych takich jak angiogenina i LIF zdecydowanie wspierają koncepcję, że aktywne reakcje zapalne układu nerwowego oraz sieć licznych cytokin uczestniczą prawdopodobnie w mechanizmach mediowanych przez układ odpornościowy w CNS pacjentów z autyzmem. Cytokiny te odgrywają ważne role w procesach mediowanych przez układ odpornościowy, zaś ich obecność w CSF pacjentów autystycznych może odzwierciedlać trwały etap reakcji zapalnych związanych prawdopodobnie z aktywacją komórek glejowych i/lub uszkodzeniem neuronów. Przyczyny relatywnie większego wzrostu tych cytokin w CSF w porównaniu z mózgiem są nieznane. Możliwe, że cytokiny pochodzą ze źródeł komórek glejowych i nerwowych, jak wykazała przeprowadzona przez nas ocena immunocytochemiczna. Różnice zaobserwowane w cytokinach w CSF w porównaniu z mózgiem mogły pochodzić z innych źródeł produkcji, takich jak opony miękkie lub splot naczyniówkowy bądź mogą oznaczać przewlekłe podwyższenie cytokin w wyniku zatrzymania w rozwoju neurologicznym, ponieważ niektóre cytokiny są zwykle podwyższone w okresie rozwoju neurologicznego. Jako że CSF łatwo jest pobrać do badań klinicznych, profile cytokin w CSF mogą być w przyszłości użyteczne w diagnozowaniu, charakteryzowaniu i śledzeniu przebiegu klinicznego zaburzeń ze spektrum autyzmu.
Wnioski
Całość naszych badań wskazuje, że reakcje komórek glejowych w postaci wrodzonej odpowiedzi immunologicznej pełnią ważną rolę w mechanizmach związanych z dysfunkcją neuronów w autyzmie oraz że móżdżek jest ogniskiem aktywnego i przewlekłego procesu zapalnego układu nerwowego u pacjentów chorych na autyzm. Obecność chemokin prozapalnych takich jak MCP-1, a także cytokin przeciwzapalnych jak TGF-β1 wspiera koncepcję, że w autyzmie występuje przewlekły stan aktywacji konkretnej cytokiny. Hipotezę tę wspiera również nasze odkrycie wyraźnego wzrostu w większej grupie cytokin w CSF, które zwykle uczestniczą w szlakach zapalnych. W świetle heterogeniczności objawów klinicznych i prawdopodobnych przyczyn autyzmu, obecność zmian zapalnych w układzie nerwowym wśród zbadanych przez nas przypadków wskazuje, że może to być częsty mechanizm patogeniczny u niektórych pacjentów z autyzmem. Ponieważ podłoże genetyczne gospodarza ma wpływ na odpowiedzi neuroimmunologiczne, rola stanu zapalnego układu nerwowego w kontekście czynników genetycznych i innych czynników decydujących o fenotypie autyzmu, pozostaje istotną kwestią, którą należy zbadać. Jako że proces zapalny układu nerwowego wydaje się mieć związek z trwałym i przewlekłym mechanizmem dysfunkcji CNS, potencjalne interwencje terapeutyczne powinny koncentrować się na kontrolowaniu jego szkodliwych skutków (utrzymując jednocześnie jego działanie naprawcze) i w ten sposób ostatecznie zmodyfikować przebieg kliniczny autyzmu.
Niniejsze badanie zostało sfinansowane z dotacji Cure Autism Now Foundation (C.A.P.), Autism Research Foundation (A.W.Z.), NIH (National Institute of Drug Abuse, K08DA016160, C.A.P.), dr Barry i Renee Gordon oraz anonimowemu darczyńcy.
Jesteśmy wdzięczni dr J. Pickettowi, Autism Tissue Program i bankom mózgów Uniwersytetu Harvard, Uniwersytetu w Miami i Uniwersytetu w Maryland za ofiarowanie tkanek mózgu. Dziękujemy dr S.L. Connorsowi, C. Eberhartowi i G. Pradilli, Jr. za ich pomocne komentarze, dr B. Paulowi Morganowi za przekazanie przeciwciała anty-C9neo, dr D. Irani za przekazanie próbek kontrolnych CSF oraz dr D. McClelanowi za pomoc redakcyjną.
Zobacz na: Zapalenie mózgu cechą autyzmu, wykazują wyniki badania z Johns Hopkins University
Zaburzenia mitochondrialne u osób z autyzmem
Szczepionki i zapalenie mózgu – dr Harold E. Buttram i Catherine J. Frompovich
Tkanki mózgowe wykorzystywane w badaniach nad autyzmem, zniszczone w wyniku awarii mroźni szpitalnej
Aluminium w mózgu osób autystycznych – prof. Chris Exley
Nano-cząsteczki metali niszczą DNA mózgu – Trinity College w Dublinie
Autyzm i Szczepionki na Świecie – Kalendarze szczepień, wskaźniki występowania autyzmu i śmiertelność poniżej 5 roku życia
Czy chińscy naukowcy odkryli brakujący fragment układanki autyzmu? – J.B. Handley
Niebezpieczeństwa nadmiernych szczepień w trakcie rozwoju mózgu – dr Russell L. Blaylock
Szczepionki nie powodują autyzmu – Marcella Piper-Terry
Omówienie hipotezy przyczyn powstawania krzywych uśmiechów i asymetrii twarzy – Forrest Maready
Nowe dane porównujące szczepionych z nieszczepionymi: wskaźnik występowania autyzmu u nieszczepionych wskazuje 1 na 715, a częściowo szczepionych 1 na 440 – Jefferey Jaxen
Bibliografia:
1. Rapin I. Autism. N Engl J Med 1997;337:97–104.
2. Lord C, Cook EH, Leventhal BL, et al. Autism spectrum disorders. Neuron 2000;28:355–363.
3. Rapin I, Katzman R. Neurobiology of autism. Ann Neurol 1998;43:7–14.
4. Newschaffer CJ, Fallin D, Lee NL. Heritable and nonheritable risk factors for autism spectrum disorders. Epidemiol Rev 2002; 24:137–153.
5. Fombonne E. Epidemiological surveys of autism and other pervasive developmental disorders: an update. J Autism Dev Disord 2003;33:365–382.
6. Bertrand J, Mars A, Boyle C, et al. Prevalence of autism in a United States population: the Brick Township, New Jersey, investigation. Pediatrics 2001;108:1155–1161.
7. Yeargin-Allsopp M, Rice C, Karapurkar T, et al. Prevalence of autism in a US metropolitan area. JAMA 2003;289:49 –55.
8. Folstein SE, Rosen-Sheidley B. Genetics of autism: complex aetiology for a heterogeneous disorder. Nat Rev Genet 2001;2: 943–955.
9. Korvatska E, Van de WJ, Anders TF, et al. Genetic and immunologic considerations in autism. Neurobiol Dis 2002;9: 107–125.
10. Lipkin WI, Hornig M. Microbiology and immunology of autism spectrum disorders. Novartis Found Symp 2003;251: 129–143.
11. Kemper TL, Bauman M. Neuropathology of infantile autism. J Neuropathol Exp Neurol 1998;57:645– 652.
12. Bailey A, Luthert P, Dean A, et al. A clinicopathological study of autism. Brain 1998;121:889 –905.
13. Bauman ML, Kemper TL. The neuropathology of the autism spectrum disorders: what have we learned? Novartis Found Symp 2003;251:112–122.
14. Licinio J, Alvarado I, Wong ML. Autoimmunity in autism. Mol Psychiatry 2002;7:329.
15. Gupta S, Aggarwal S, Rashanravan B, et al. Th1- and Th2-like cytokines in CD4 and CD8 T cells in autism. J Neuroimmunol 1998;85:106 –109.
16. Singh VK, Warren R, Averett R, et al. Circulating autoantibodies to neuronal and glial filament proteins in autism. Pediatr Neurol 1997;17:88 –90.
17. Vojdani A, Campbell AW, Anyanwu E, et al. Antibodies to neuron-specific antigens in children with autism: possible crossreaction with encephalitogenic proteins from milk, Chlamydia pneumoniae and Streptococcus group A. J Neuroimmunol 2002; 129:168 –177.
18. Jyonouchi H, Sun S, Le H. Proinflammatory and regulatory cytokine production associated with innate and adaptive immune responses in children with autism spectrum disorders
and developmental regression. J Neuroimmunol 2001;120: 170–179.
19. Dalton P, Deacon R, Blamire A, et al. Maternal neuronal antibodies associated with autism and a language disorder. Ann Neurol 2003;53:533–537.
20. Comi AM, Varsou A, Heyes MP, et al. Quinolinic acid and neopterin in children with autism: an analysis of cerebrospinal fluid. Ann Neurol 1999;46:528 –529.
21. Zimmerman AW. Commentary: immunological treatments for autism: in search of reasons for promising approaches. J Autism Dev Disord 2000;30:481– 484.
22. Guerin P, Lyon G, Barthelemy C, et al. Neuropathological study of a case of autistic syndrome with severe mental retardation. Dev Med Child Neurol 1996;38:203–211.
23. Lord C, Pickles A, McLennan J, et al. Diagnosing autism: analyses of data from the Autism Diagnostic Interview. J Autism Dev Disord 1997;27:501–517.
24. Scahill L, Lord C. Subject selection and characterization in clinical trials in children with autism. CNS Spectr 2004;9:22–32.
25. Gundersen HJ, Bendtsen TF, Korbo L, et al. Some new, simple and efficient stereological methods and their use in pathological research and diagnosis. APMIS 1988;96:379 –394.
26. Vehmas AK, Kawas CH, Stewart WF, et al. Immune reactive cells in senile plaques and cognitive decline in Alzheimer’s disease. Neurobiol Aging 2003;24:321–331.
27. Huang RP. Cytokine protein arrays. Methods Mol Biol 2004; 278:215–232.
28. Lin Y, Huang R, Chen LP, et al. Profiling of cytokine expression by biotin-labeled-based protein arrays. Proteomics 2003;3: 1750–1757.
29. Storch MK, Piddlesden S, Haltia M, et al. Multiple sclerosis: in situ evidence for antibody- and complement-mediated demyelination. Ann Neurol 1998;43:465– 471.
30. Aloisi F. Immune function of microglia. Glia 2001;36: 165–179.
31. Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci 1996;19:312–318.
32. Bauer J, Rauschka H, Lassmann H. Inflammation in the nervous system: the human perspective. Glia 2001;36:235–243.
33. Medzhitov R, Janeway CA Jr. Innate immune recognition and control of adaptive immune responses. Semin Immunol 1998; 10:351–353.
34. Aloisi F. The role of microglia and astrocytes in CNS immune surveillance and immunopathology. Adv Exp Med Biol 1999; 468:123–133.
35. McGeer PL, Kawamata T, Walker DG, et al. Microglia in degenerative neurological disease. Glia 1993;7:84 –92.
36. Teismann P, Tieu K, Cohen O, et al. Pathogenic role of glial cells in Parkinson’s disease. Mov Disord 2003;18:121–129.
37. Henkel JS, Engelhardt JI, Siklos L, et al. Presence of dendritic cells, MCP-1, and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord tissue. Ann Neurol 2004;55: 221–235.
38. Glass JD, Wesselingh SL. Microglia in HIV-associated neurological diseases. Microsc Res Tech 2001;54:95–105.
39. Gartner S. HIV infection and dementia. Science 2000;287: 602–604.
40. Banati RB, Gehrmann J, Schubert P, et al. Cytotoxicity of microglia. Glia 1993;7:111–118.
41. Gebicke-Haerter PJ. Microglia in neurodegeneration: molecular aspects. Microsc Res Tech 2001;54:47–58.
42. Auld DS, Robitaille R. Glial cells and neurotransmission: an inclusive view of synaptic function. Neuron 2003;40:389–400.
43. Fields RD, Stevens-Graham B. New insights into neuron-glia communication. Science 2002;298:556 –562.
44. Nedergaard M, Ransom B, Goldman SA. New roles for astrocytes: redefining the functional architecture of the brain. Trends Neurosci 2003;26:523–530.
45. Allan SM, Rothwell NJ. Cytokines and acute neurodegeneration. Nat Rev Neurosci 2001;2:734 –744.
46. Nguyen MD, Julien JP, Rivest S. Innate immunity: the missing link in neuroprotection and neurodegeneration? Nat Rev Neurosci 2002;3:216 –227.
47. Rezaie P, Male D. Colonisation of the developing human brain and spinal cord by microglia: a review. Microsc Res Tech 1999; 45:359 –382.
48. Wierzba-Bobrowicz T, Kosno-Kruszewska E, Gwiazda E, et al. Major histocompatibility complex class II (MHC II) expression during the development of human fetal cerebral occipital lobe, cerebellum, and hematopoietic organs. Folia Neuropathol 2000; 38:111–118.
49. McGeer PL, McGeer EG. Innate immunity, local inflammation, and degenerative disease. Sci Aging Knowledge Environ 2002;2002:re3.
50. Courchesne E, Yeung-Courchesne R, Press GA, et al. Hypoplasia of cerebellar vermal lobules VI and VII in autism. N Engl J Med 1988;318:1349 –1354.
51. Courchesne E. Neuroanatomic imaging in autism. Pediatrics 1991;87:781–790.
52. Carper RA, Courchesne E. Inverse correlation between frontal lobe and cerebellum sizes in children with autism. Brain 2000; 123:836–844.
53. Kinnear KJ. Purkinje cell vulnerability and autism: a possible etiological connection. Brain Dev 2003;25:377–382.
54. Leonard EJ, Yoshimura T. Human monocyte chemoattractant protein-1 (MCP-1). Immunol Today 1990;11:97–101.
55. Mahad DJ, Ransohoff RM. The role of MCP-1 (CCL2) and CCR2 in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE). Semin Immunol 2003;15:23–32.
56. Kelder W, McArthur JC, Nance-Sproson T, et al. Betachemokines MCP-1 and RANTES are selectively increased in cerebrospinal fluid of patients with human immunodeficiency virus- associated dementia. Ann Neurol 1998;44:831– 835.
57. Losy J, Zaremba J. Monocyte chemoattractant protein-1 is increased in the cerebrospinal fluid of patients with ischemic stroke. Stroke 2001;32:2695–2696.
58. Eugenin EA, D’Aversa TG, Lopez L, et al. MCP-1 (CCL2) protects human neurons and astrocytes from NMDA or HIVtat- induced apoptosis. J Neurochem 2003;85:1299 –1311.
59. Peterson KE, Errett JS, Wei T, et al. MCP-1 and CCR2 contribute to non-lymphocyte-mediated brain disease induced by Fr98 polytropic retrovirus infection in mice: role for astrocytes in retroviral neuropathogenesis. J Virol 2004;78:6449–6458.
60. Meng SZ, Oka A, Takashima S. Developmental expression of monocyte chemoattractant protein-1 in the human cerebellum and brainstem. Brain Dev 1999;21:30 –35.
61. Letterio JJ, Roberts AB. Regulation of immune responses by TGF-beta. Annu Rev Immunol 1998;16:137–161.
62. Chen W, Frank ME, Jin W, et al. TGF-beta released by apoptotic T cells contributes to an immunosuppressive milieu. Immunity 2001;14:715–725.
63. Mundy P. Annotation: the neural basis of social impairments in autism: the role of the dorsal medial-frontal cortex and anterior cingulate system. J Child Psychol Psychiatry 2003;44:793– 809.
64. Franciotta D, Martino G, Zardini E, et al. Serum and CSF levels of MCP-1 and IP-10 in multiple sclerosis patients with acute and stable disease and undergoing immunomodulatory
therapies. J Neuroimmunol 2001;115:192–198.